

| Journal of Neurology Research, ISSN 1923-2845 print, 1923-2853 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Neurol Res and Elmer Press Inc |
| Journal website http://www.neurores.org |
Original Article
Volume 9, Number 4-5, October 2019, pages 60-64
Update on an Asian Indian Family With Apparent Autosomal Recessive Charcot-Marie-Tooth Disease Caused by a Mutation in the HSPB1 Gene
Leema Reddy Peddareddygaria, b, Kinsi Oberoia, c, Raji P. Grewald, e
aThe Neurogenetics Foundation, Cranbury, NJ, USA
bDynamic Biologics Inc., 7 Deer Park Drive, Monmouth Junction, NJ, USA
cClarivate Analytics, Life Sciences Division, 1500 Spring Garden Street, Philadelphia, PA, USA
dNeuroscience Institute, Saint Francis Medical Center, 601 Hamilton Avenue, Trenton, NJ, USA
eCorresponding Author: Raji P. Grewal, Neuroscience, Seton Hall University/Saint Francis Medical Center, Trenton, NJ 08629, USA
Manuscript submitted July 10, 2019, accepted July 31, 2019
Short title: Genetic Neuropathy Caused by HSPB1 Gene Mutation
doi: https://doi.org/10.14740/jnr547
| Abstract | ▴Top |
Background: We described an Asian Indian family with a genetic neuropathy previously in the Journal of Neurology Research, 2012. In that publication, we speculated that a deletion mutation in the PRX gene may have contributed to the development of the neuropathy. In this family, there is significant phenotypic variability which created difficulties establishing the mode of transmission which appeared to be autosomal recessive. We now present our updated analysis with additional clinical and genetic data.
Methods: We obtained clinical and phenotype data on additional members of this family. We performed whole exome sequencing on the index patient and targeted genotyping of other members of the family.
Results: Our updated analysis establishes the pattern of inheritance of this neuropathy as autosomal dominant and caused by a mutation in the HSPB1 gene, R140G. The R140G mutation has been previously reported in a number of unrelated families originating from Gujarat, the same Indian state as the subjects of this study.
Conclusions: The collective genetic analysis of this mutation in the Gujarati families suggests the presence of a founder effect of the R140G mutation in this population. Our investigation of this family demonstrates the capacity of next generation sequencing in facilitating the ability to make a specific genetic diagnosis.
Keywords: Charcot-Marie-Tooth disease; Adult onset; HSPB1 gene; R140G mutation
| Introduction | ▴Top |
Charcot-Marie-Tooth (CMT) disease represents a clinically and genetically heterogeneous group of disorders affecting both motor and sensory nerves of the peripheral nervous system [1].
In 2012, in this journal, we reported a patient carrying a novel deletion mutation in the PRX gene which we speculated could have contributed to the development of the neuropathy in this family [2]. We have followed this family now for more than 10 years. During this time, the index patient has further deteriorated and two of his sons have become symptomatic. Interestingly, the remaining son, who is the only one carrying the PRX gene deletion, has remained asymptomatic suggesting this is not the pathogenic mutation causing the neuropathy. In this report, we present our updated clinical and genetic analysis of this family.
| Materials and Methods | ▴Top |
Clinical data
This index patient (Fig. 1, patient III-5) was then a 59-year-old Asian Indian when he first presented for an evaluation. The details of his history, neurological examination and electromyography (EMG) testing are described in detail as “Case 2” in the prior report. Briefly, he had been diagnosed with a peripheral neuropathy with predominantly motor fiber involvement. He was significantly disabled from this illness which began when he was in his early 40s and progressed so that he required bilateral foot orthotic devices. He had no symptoms of cranial nerve or autonomic system dysfunction and denied complaints of sensory loss or neuropathic pain. Since the initial visit, the patient, who is now 73 years old has clinically deteriorated and now requires a walker or a wheelchair for mobility. His complaints continue to be those of increased weakness of his arms and legs without sensory complaints or features to indicate autonomic nervous system involvement. There are no abnormalities of the mental status or the cranial nerve examination. His neurological examination shows distal weakness of his arms and legs associated with atrophy. The weakness is more severe in his legs Medical Research Council (MRC) Grade 2-3/5. His reflexes are preserved and his plantar responses are flexor. Sensory examination shows a mild decrease in light touch distally in his feet with preservation of proprioception and vibratory sense. He cannot walk without an assistive device.
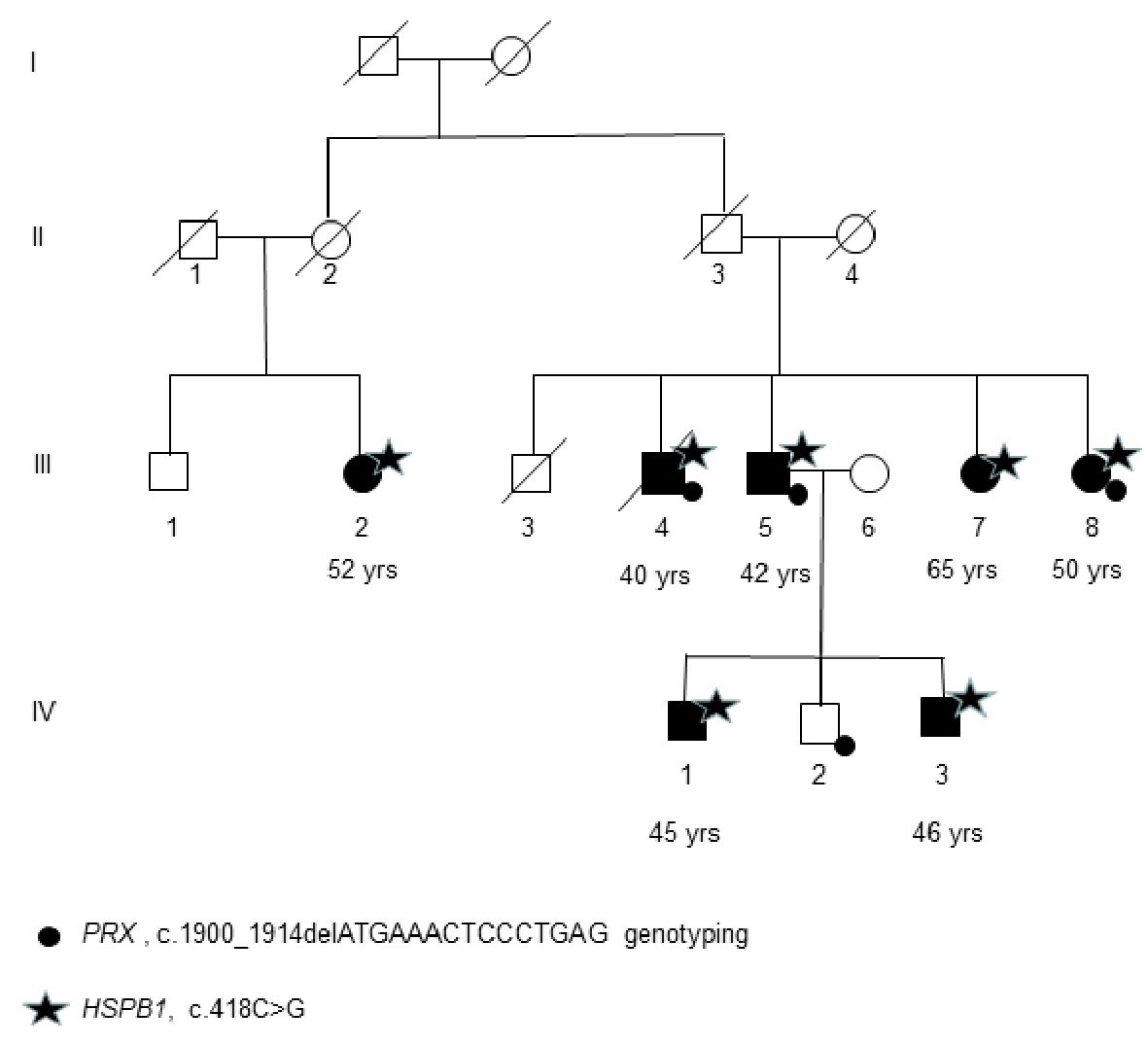 Click for large image | Figure 1. Pedigree of the family with adult-onset Charcot-Marie-Tooth disease. The squares indicate males, circles indicate females, and dark fill indicates symptomatic individuals. The black dot indicates individuals with the deletion mutation in PRX gene and the black star indicates individuals carrying HSPB1 gene variant. The number below the symbols represents the approximate age of onset of affected individuals. |
He has three sons whose ages ranged from 34 to 45 years at the time of the original report and who, at that time, were all asymptomatic (Fig. 1). We had examined individual IV-1 at age 40 years and both his neurological exam and EMG study were normal. At age 45 years he noted some difficulties with his gait and ultimately came for a re-evaluation 2 years after the onset of these symptoms. Neurological examination revealed normal tests of his mental status, cranial nerves and sensation. He had normoactive reflexes with flexor plantar responses. Power testing revealed distal weakness of foot dorsiflexion, eversion and inversion (MRC 4/5), and his gait demonstrated bilateral foot drop. A repeat EMG indicates a neurogenic process affecting the muscles innervated by lumbosacral myotomes L5-S1 with acute and chronic neurogenic changes. Sensory nerve action potentials (SNP) in the sural and superficial peroneal nerves were normal. Another son (IV-3) had previously declined to participate in the study, however, at age 46 years he sought neurological evaluation for complaints of limping without back pain or sensory loss. His examination at age 51 years demonstrated distal weakness MRC 4/5 in his legs with hyperactive reflexes at the patella and ankles and flexor plantar responses. A review of an EMG done 2 years earlier showed the presence of a neurogenic process with acute and chronic changes in the muscles innervated by the L4-S1 myotomes. The presence of normal SNAPs in both sural and superficial nerves was noted. A third son (IV-2) had no complaints of weakness or imbalance, and when examined at age 34 years, he had a normal neurological examination. Now he remains asymptomatic at age 41 years.
The index patient’s brother (III-4) developed symptoms of weakness and imbalance when he was 40 years old. Over the ensuing 40 years, these symptoms progressed and he became wheelchair bound. A neurological examination performed was consistent with a severe motor neuropathy. Since the publication of our report, his neurological status worsened and he became non-ambulatory. He died from the complications of cancer in his late 80s. He has three sons ranging in age from 52 to 59 years, who remain asymptomatic by history.
The index patient has two sisters. An older one (III-7) now age 72 years was initially designated as unaffected. However she has an abnormal gait that began about 6 years earlier and she now walks with a cane suggesting she may be affected. The other sister, (III-8), now age 67 years has leg weakness and a “funny walk” which began in her 50s. She was designated as affected in the prior report and her abnormal gait has progressed. She is still ambulatory with the use of a cane.
When the index patient was initially evaluated, it was recorded that his father had died from cancer in his 50s and was asymptomatic. His mother died in her 80s without neurological complaints. Further information has been obtained indicating that a female cousin (III-2) on the paternal side of the family is affected with complaints of weakness and imbalance which began at approximately age 52 years. This study was conducted in compliance with the ethical standards of the responsible institution on human subjects as well as with the Helsinki Declaration.
Genetic analysis
Since the initial report, we proceeded with whole exome sequencing on the index patient and targeted genotyping of other members of the family. Following Institutional Review Board approved policies and procedures, a blood sample was obtained from all members of the family who consented for the study, and genomic DNA was extracted using whole blood extraction kit (Qiagen, Puregene). Whole exome sequencing was performed by HISeq2500 using paired-end (2 × 100) protocol, Illumina raw data processing and SureSelect (SureSelect Human All Exon Target Enrichment System, Agilent Technologies) exome capture methods. The nucleotide-level variation analysis of the exome sequence data was performed using the DNA nexus platform. We mapped paired-end-reads to the reference human genomes (UCSC NCBI37/hg19) using Burrows-Wheeler Alignment tool (BWA). Single nucleotide polymorphisms and indels were called by GATK-lite variant caller, and the Ensambl VEP tool was used to annotate and filter variants.
We developed a polymerase chain reaction (PCR) to identify the HSPB1 gene variant, and all available members in the family were then screened. The PCR was performed with a final volume for 50 µL, containing 40 ng of genomic DNA, 45 µL of platinum mix (22 U/mL recombinant Taq DNA polymerase complexed with Platinum Taq antibody, 22 mM Tris-HCl (pH 8.4), 55 mM KCl, 1.65 mM MgCl2, 220 µM dGTP, 220 µM dATP, 220 µM dTTP, 220 µM dCTP, and stabilizers) and 0.4 µM of each primer (forward: 5’-CCGCAGTCTGATTTCCCTCT-3’ and reverse: 5’-GAGGAAAGGCAAGCGTTACA-3’). The PCR cycling parameters used includes initial denaturation 95 °C for 7 min followed by 34 cycles (95 °C for 30 s, 60 °C for 30 s, 72 °C for 30 s) with a final extension at 72 °C for 7 min. This PCR product (188 bp) was subjected to digestion with BstUI enzyme for 2 h at 60 °C and resolved by gel electrophoresis on a 4% agarose gel. The wild-type allele with the restriction site intact results in two fragments, 84 bp and 104 bp while the mutant allele remains undigested due to loss of restriction site resulting in a single 188 bp product.
| Results | ▴Top |
Whole exome sequence analysis identified two variants, a 15 bp deletion mutation in the Periaxin gene, rs575492732 (chr19: 40902345-40902359delCTCAGGGAGTTTCAT, c.1900-1914delATGAAACTCCCTGAG, p.Met634-Glu638del) and a single nucleotide variant in heat shock 27 kDa protein 1 (HSPB1) gene, rs121909112 (chr7:75933172C>G, c.418C>G, p.Arg140Gly).
The results of sequencing the HSPB1 gene c.418C>G variant using PCR is shown in Figure 2a and b, which show the results of sequencing the PRX gene 15 bp deletion variant in this family [2]. Individuals III-4, III-5 and III-8 inherited both HSPB1 and the PRX gene mutation while others inherited either the HSPB1 (III-2, III-7, IV-1 and IV-3) or the PRX gene (IV-2) variant (Fig. 1).
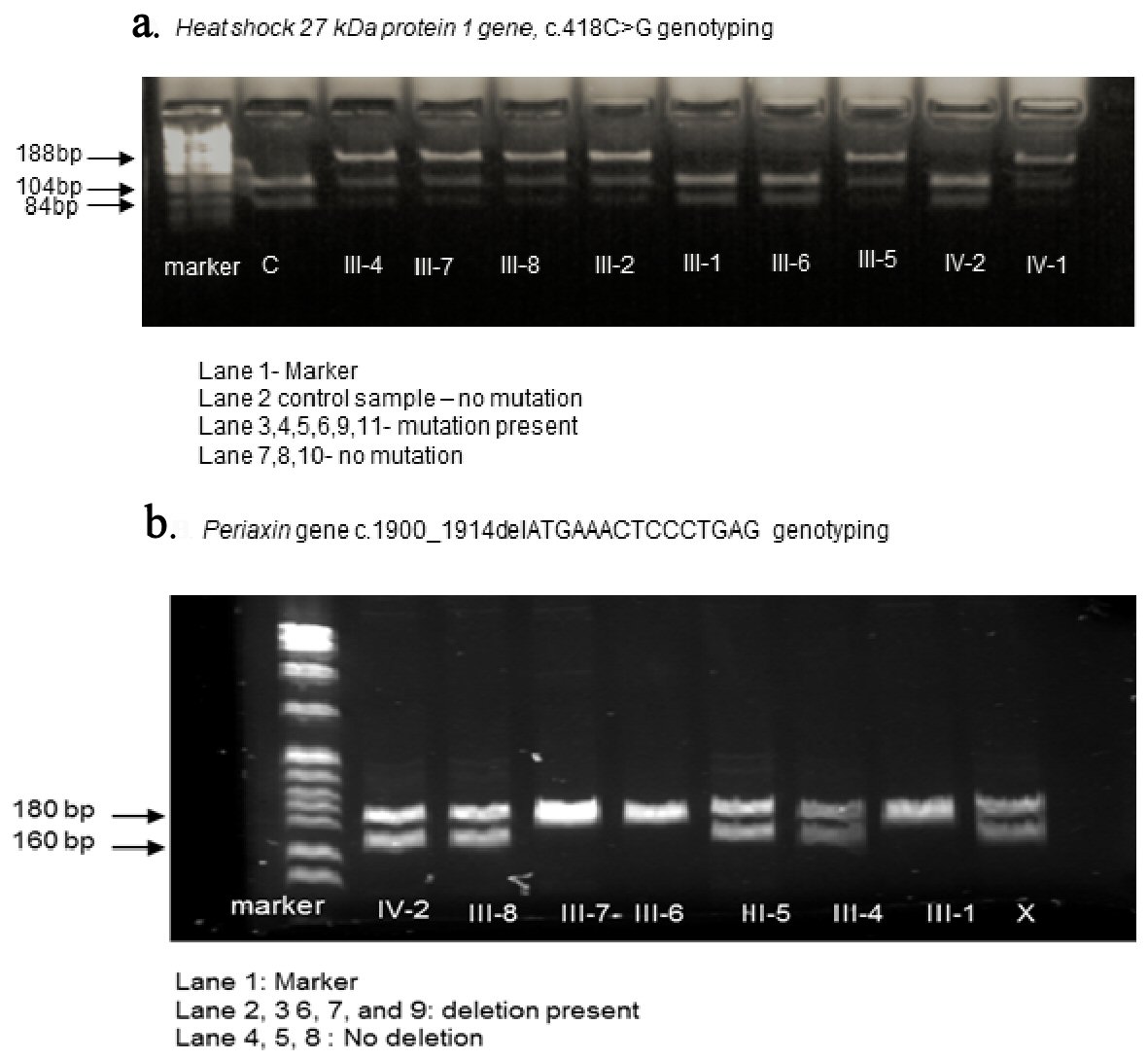 Click for large image | Figure 2. Genotyping analysis of the HSPB1 (a) and PRX (b) genes in this family. PCR products are resolved on a 4% agarose gel. PCR: polymerase chain reaction. |
| Discussion | ▴Top |
In our initial study of this family, the presentation appeared to be that of a recessive disease. However, with the information now available, it is clear that the mode of transmission of this neuropathy is autosomal dominant. Our updated genetic analysis confirms that it is caused by the R140G mutation in the HSPB1 gene.
Mutations in the HSPB1 gene have been reported to cause autosomal dominant, autosomal recessive or sporadic distal HMN/CMT type 2F [3, 4]. Although it appeared that the index patient’s father was not affected raising the possibility of a sporadic mutation, given the genotype of patient III-2 it is clear that he was an obligate carrier. He may have been asymptomatic or, perhaps minimally symptomatic at the time of his death.
The specific mutation detected in this family, R140G, was originally reported in three families of Asian Indian descent. In one patient it followed an autosomal dominant pattern of inheritance while in other two it was reported as a sporadic mutation [4]. Interestingly, a second publication from the same institution and group discussed the phenotype and natural history of patients with mutations in the HSPB gene. In this follow-up study, five families are reported of Asian Indian descent, four from the state of Gujarat [5]. In a subsequent publication, again by the same group, it is indicated that all five are from Gujarat and all are recorded as demonstrating an autosomal dominant form of transmission of the R140G mutation [6]. This group also reported a sixth patient and family with a homozygous R140G mutation. Remarkably this patient’s parents had no neurological complaints but an examination in their 80s revealed MRC 4/5 weakness of arms and legs indicating disease manifestation of these obligate carriers. A seventh patient carrying this same mutation has been reported from Western India which we suspect is of Gujarati descent [7]. Our study reports the eighth family of Asian Indian/Gujarati origin carrying this specific mutation. All of these families demonstrate that significant phenotypic variability can occur in carriers of the R140G mutation which can create confusion about the mode of inheritance.
Collectively, all of these families suggest a founder effect for the R140G mutation in the Gujarati population. It may be noted that the mutation is not exclusive to this population as it has been reported in two patients of Japanese origin also with autosomal dominant pattern of inheritance [8, 9]. In our family, it is possible that there is an interaction between the PRX deletion and the R140G mutation resulting in a more severe phenotype in those individuals carrying both these mutations. Our analysis shows that carrying only the PRX deletion does not result in development of a neuropathy. The two male siblings (III-4, III-5) with the digenic inheritance of both of these mutations developed symptoms early and more severe than other members of the family. There is biological plausibility for the concept of the PRX gene as a potential genetic modifier as it is necessary not only for myelination but for axon integrity [10]. However, it must be noted that the sister carrying both mutations (III-8) did not develop the neuropathy early or with the degree of severity as her siblings. Therefore, there must be other genetic modifiers that result in the phenotypic variability observed in these families.
Our analysis of this family provides the value of long-term follow-up of patients and families which, in this case, enabled documentation of an autosomal dominant form of transmission rather than autosomal recessive or a sporadic mutation. In addition, our study provides a demonstration of the clinical utility of next generation sequencing in the analysis of patients with genetic disorders. Without the capacity to perform whole exome sequencing at a reasonable cost, we would not have been able to identify the mutation in this family. Advances in genetics will continue to improve our ability to identify the mutations that cause genetic disorders and hopefully, facilitate the development of effective therapies.
Acknowledgments
We acknowledge the support of the Neurogenetics Foundation and the family members for their cooperation.
Financial Disclosure
None to declare.
Conflict of Interest
The authors declare that there is no conflict of interests regarding the publication of this paper.
Informed Consent
This study was approved by the local Institutional Review Board (IRB). Informed consents were obtained.
Author Contributions
All authors contributed to initial drafts of this publication, RPG and LRP revised later drafts. All authors reviewed and approved the final draft. The neurological examinations were performed by RPG. PLR and KO were involved in generation and analysis of the genetic data. RPG was involved in supervision of all aspects of the study.
| References | ▴Top |
- Skre H. Genetic and clinical aspects of Charcot-Marie-Tooth's disease. Clin Genet. 1974;6(2):98-118.
doi pubmed - Peddareddygari LR, Sobol I, Pillai HB, Ito M, Batish SD, Grewal RP. Analysis of association of deletion in the repeat region of the periaxin gene with late onset motor neuropathy. J Neurol Res. 2012;2(6):235-243.
doi - Evgrafov OV, Mersiyanova I, Irobi J, Van Den Bosch L, Dierick I, Leung CL, Schagina O, et al. Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat Genet. 2004;36(6):602-606.
doi pubmed - Houlden H, Laura M, Wavrant-De Vrieze F, Blake J, Wood N, Reilly MM. Mutations in the HSP27 (HSPB1) gene cause dominant, recessive, and sporadic distal HMN/CMT type 2. Neurology. 2008;71(21):1660-1668.
doi pubmed - Rossor AM, Morrow JM, Polke JM, Murphy SM, Houlden H, Inc R, Laura M, et al. Pilot phenotype and natural history study of hereditary neuropathies caused by mutations in the HSPB1 gene. Neuromuscul Disord. 2017;27(1):50-56.
doi pubmed - Bugiardini E, Rossor AM, Lynch DS, Swash M, Pittman AM, Blake JC, Hanna MG, et al. Homozygous mutation in HSPB1 causing distal vacuolar myopathy and motor neuropathy. Neurol Genet. 2017;3(4):e168.
doi pubmed - Khadilkar SV, Patil ND, Kadam ND, Mansukhani KA, Patel BA. Clinico-Electrophysiological and genetic overlaps and magnetic resonance imaging findings in Charcot-Marie- tooth disease: A Pilot Study from Western India. Ann Indian Acad Neurol. 2017;20(4):425-429.
doi pubmed - Tanabe H, Higuchi Y, Yuan JH, Hashiguchi A, Yoshimura A, Ishihara S, Nozuma S, et al. Clinical and genetic features of Charcot-Marie-Tooth disease 2F and hereditary motor neuropathy 2B in Japan. J Peripher Nerv Syst. 2018;23(1):40-48.
doi pubmed - Yoshimura A, Yuan JH, Hashiguchi A, Ando M, Higuchi Y, Nakamura T, Okamoto Y, et al. Genetic profile and onset features of 1005 patients with Charcot-Marie-Tooth disease in Japan. J Neurol Neurosurg Psychiatry. 2019;90(2):195-202.
doi pubmed - Kim S, Maynard JC, Sasaki Y, Strickland A, Sherman DL, Brophy PJ, Burlingame AL, et al. Schwann cell O-GlcNAc glycosylation is required for myelin maintenance and axon integrity. J Neurosci. 2016;36(37):9633-9646.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Neurology Research is published by Elmer Press Inc.