

| Journal of Neurology Research, ISSN 1923-2845 print, 1923-2853 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Neurol Res and Elmer Press Inc |
| Journal website http://www.neurores.org |
Original Article
Volume 2, Number 6, December 2012, pages 235-243
Analysis of Association of Deletion in the Repeat Region of the Periaxin Gene With Late Onset Motor Neuropathy
Leema Reddy Peddareddygaria, b, Ilya Sobolc, Harish B Pillaia, Masamichi Itod, Sat D. Batishd, Raji P. Grewalc, e
aThe Neurogenetics Foundation, Cranbury, New Jersey, USA
bClinnovo Research Labs, KhanMet, Hitech city, Hyderabad, Andhra Pradesh, India
cNeuroscience Institute, Saint Francis Medical Center, 601 Hamilton Avenue, Trenton, New Jersey, USA
dAthena Diagnostics, Inc., Worcester, Massachusetts, USA
eCorresponding author: Raji P. Grewal, Seton Hall University/Saint Francis Medical Center, 601 Hamilton Ave, Trenton, NJ, USA
Manuscript accepted for publication December 11, 2012
Short title: Deletion of the Periaxin Gene
doi: https://doi.org/10.4021/jnr154w
| Abstract | ▴Top |
Background: Charcot-Marie-Tooth Disease (CMT) represents a heterogeneous group of disorders affecting the peripheral nervous system. The protein encoded by the periaxin (PRX) gene has a functional role in the maintenance of peripheral nerve myelin. Mutations in the PRX gene have been identified in autosomal recessive forms of Dejerine-Sottas neuropathy and Charcot Marie Tooth disease type 4F. Both of these conditions are characterized by early onset of a demyelinating neuropathy.
Methods: We report two unrelated patients, a Caucasian American and an Asian American patient with late onset progressive neuropathy. We also ascertained family members of the Asian American patient for clinical and genetic evaluation.
Results: We report heterozygous deletions in the repeat region of the PRX gene; the Caucasian patient developed symptoms as a teenager and has a 78 base pair deletion. The Asian patient became symptomatic in his forties and has a 15 base pair deletion and the same deletion was also identified in two other siblings with neuropathy. Both deletion mutations involve the repeat region in exon 7 of the PRX gene. In both patients, electrophysiological analysis demonstrates a predominantly motor neuropathy with demyelinating features.
Conclusions: This is the first report of deletions involving the repeat region in exon 7 of the PRX gene. We present epidemiological data and discuss the possible significance of the association of these deletion mutations with neuropathy.
Keywords: Charcot-Marie-Tooth Disease; Adult onset; Periaxin gene; Repeat region; Deletion mutation; Axonal polyneuropathy
| Introduction | ▴Top |
Charcot-Marie-Tooth (CMT) disease represents a clinically and genetically heterogeneous group of disorders affecting both motor and sensory nerves of the peripheral nervous system. CMT disease, alternatively known as hereditary motor sensory neuropathy (HMSN), is a common neurological disorder with a frequency of about 1/2,500 [1]. Traditionally, CMT disease has been classified by clinical, electrophysiological and genetic criteria [2, 3]. The genetic classification has been based upon the mode of transmission which can be autosomal dominant (AD), autosomal recessive (AR) or X-linked. Autosomal recessive CMT (ARCMT) is uncommon representing 10% of all cases of CMT [4] and, based upon electrophysiological features, can be further subdivided into demyelinating (AR CMT1 and CMT4) or axonal (ARCMT2) form. However, with the discovery of the molecular basis of many of the subtypes of ARCMT, a more specific genetic classification system has been established. CMT4 can now be sub-classified into CMT4 (A-J) and each of these ten subtypes represents a mutation in a specific gene. CMT4F is caused by mutations in the periaxin (PRX) gene [5].
Periaxin was first identified as a protein expressed by myelinating Schwann cells of the developing mammalian peripheral nervous system [6]. The L- and S-periaxin proteins are both encoded by the periaxin(PRX) gene and contain a PDZ domain named by combining the first letters of three proteins in which they were first identified. These include the post synaptic density (PSD95), Drosophila disc large tumor suppressor (DlgA), and the zonula occludens-1 proteins (ZO-1) [7, 8].The PDZ domain of L-Periaxin binds to the dystrophin-related protein, DRP2, and dystroglycan at the Schwann cell plasma membrane andfunctions to maintain the integrity and stability of the myelin sheath [9]. It was demonstrated that periaxin null mice lacking a functional periaxin develop a severe demyelinating neuropathy associated with allodynia and hyperalgesia [10]. A number of mutations have been described inPRX gene. These range from transition mutations producing substitutions of amino acids, 1-base pair (bp) and 4-bp deletions resulting in a frame shift and premature truncation of the protein and a 10-bp insertion mutation [11-16]. These periaxin mutations are all autosomal recessive, inherited in either a homozygous or compound heterozygous mutation state. These patients develop a neuropathy that is clinical and electrophysiologically characterized by early onset, little or slow progression and demyelinating features. Epidemiologically, mutations in the PRX gene have been identified in a variety of racial/ethnic groups from Lebanon, North America, Europe, Vietnam and Japan.
We report the association of deletions in the PRX gene in two unrelated patients, one a Caucasian American and other an Asian American with a late onset and predominantly motor neuropathy with demyelinating features. We also report the clinical and genetic analysis of the family members of the Asian American patient.
| Material and Methods | ▴Top |
Case 1
This is a 63-year-old Caucasian American man who began to have difficulties with his gait as a teenager. He could not run well and frequently sprained his ankles. In spite of these difficulties, he was able to serve in the military in a non-combatant role. He was ultimately referred for neurological evaluation to another institution at age 35 years and was diagnosed with Charcot-Marie-Tooth disease, the original records were not available for review. The patient does not recall having difficulties with numbness, tingling or pain. At that time, he was prescribed bilateral mid-calf ankle orthosis and has used them ever since. Over the ensuing time period he has continued to have difficulties not only with his legs but more recently, his arms. There are no complaints of numbness or tingling and no symptoms to suggest cranial nerve, mental status or the autonomic system dysfunction.
At age 49 years he was diagnosed with diabetes mellitus and treated with oral hypoglycemic medications.
Neurological examination at our institution revealed a normal mental status and cranial nerve examination. Muscle stretch reflexes were normal in the arms and the patellae and not obtained at the ankles. The plantar responses were flexor. Motor strength testing showed Grade 4/5 weakness of right wrist flexion and of the finger extensors. There was no weakness in the triceps, brachioradialis or other muscles tested in the rights arm. Strength was normal in the left arm testing both the proximal and distal musculature. In his legs, he had weakness of foot dorsiflexion and eversion (Grade 1/5), inversion and plantar flexion Grade 4/5 bilaterally. The proximal leg muscles tested were all normal including the quadriceps, iliopsoas, hamstrings, hip adductors and abductors. Sensory examination showed a mild decrease in light touch and temperature distally in his feet. Proprioception and vibration sense was normal. Cerebellar testing was normal and gait testing showed bilateral foot drop.
Family history
Family history reveals that his parents died in their 70’s with no neurological problems. No other member of the extended family is known to be affected with neuropathy including a brother and two children, both in their 20’s.
Laboratory testing
The following investigations were normal or negative: CBC and differential, comprehensive metabolic panel, serum protein electrophoresis and immunofixation, vitamin B12, folate, serology for lyme disease, HIV, hepatitis C, anti-myeloperoxidase, anti-proteinase-3, c-ANCA, p-ANCA, ANA, rheumatoid factor, anti-ganglioside antibody panel (GD1b, GD1a, GM1, G1qb).
This patient has had a number of electrophysiological studies performed over the years and the most recent is summarized in Table 1.
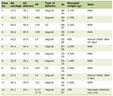 Click to view | Table 1. Nerve Conduction Studies in Case 1 |
It demonstrates the presence of severe and preferential involvement of the bilateral peroneal nerves with demyelinating features. There is relatively mild involvement of the sural and right superficial sensory nerves most likely secondary to diabetes mellitus.
This patient has been evaluated at several academic institutions and based upon his neurological examination and electrophysiologic testing has been variously diagnosed as having HNNP and multi-focal motor neuropathy.
Genetic testing
Testing for the following genes known to cause a neuropathy was performed and was negative: Cx32, MPZ, PMP22 (sequencing, deletion and duplication analysis), EGR2, NFL, GDAP1, LITAF, MFN2, SH3TC2, FIG4, LMNA, RAB7, GARS and HSPB1.
Analysis of the PRX gene showed a 78 base pair deletion from 1483-1560 affecting codon positions 495-520.
Case 2
Figure 1, individual III-5, the index patient, a 59-year-old immigrant from Gujarat, India, presented for evaluation of complaints of progressive leg weakness. At age 44 years he started to develop a gait disturbance and over the ensuing 2 - 3 years, had difficulty walking fast. At age 53, he noted muscle atrophy in his calves associated with bilateral foot drop and sought medical attention. He had no complaints of sensory loss or autonomic symptoms. He underwent an evaluation at another institution and neurological examination showed normal stretch reflexes in his arms and at the patellae. He had loss of the ankle reflexes and his plantar reflexes were flexor. He had (MRC grade 3/5) weakness of foot plantar, dorsiflexion, eversion and inversion. The strength in the proximal leg muscles was normal. In his arms, strength testing, reflex and sensory examinations were normal. The remainder of the neurological examination disclosed no abnormalities. After further investigations were completed including an electromyography (EMG), the patient was diagnosed with a motor neuropathy with a differential diagnosis of adult onset spinal muscular atrophy, poliomyelitis and spinal and bulbar muscular atrophy. Genetic testing was performed and is summarized below.
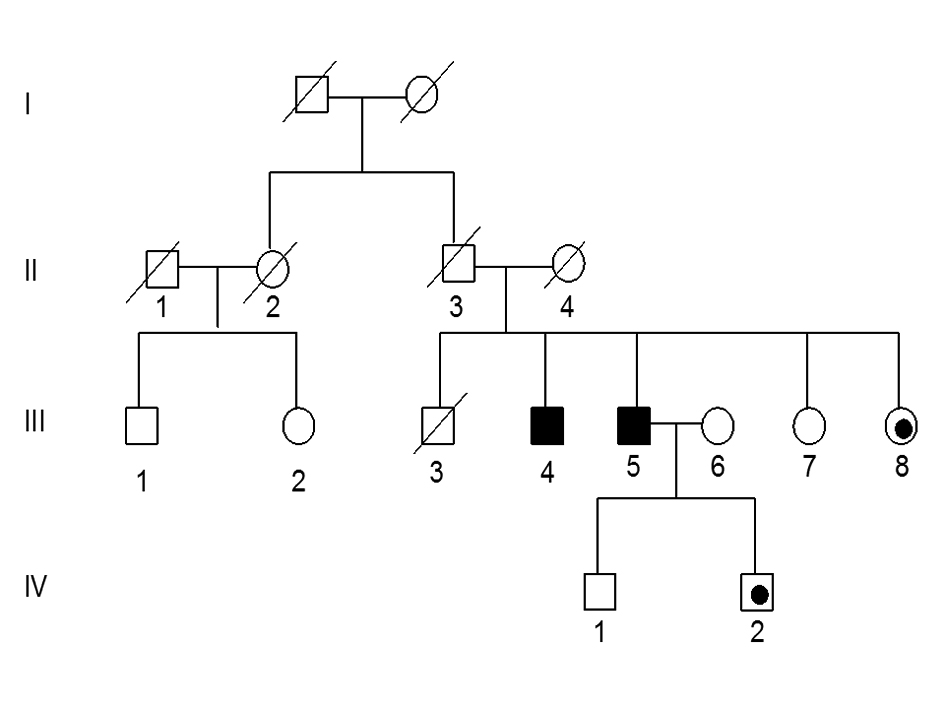 Click for large image | Figure 1. Pedigree of family with adult onset CMT disease. Squares indicate males, circle females, dark fill indicates affected individuals with deletion mutation, black dot indicates individuals with the deletion but not confirmed clinically. |
He presented for neuromuscular consultation to our institution and over the ensuing six years he developed weakness of both of his hands and progression of gait imbalance and leg weakness. He has had no symptoms of cranial nerve or autonomic system dysfunction and denied complaints of sensory loss or neuropathic pain. Neurological evaluation revealed a normal mental status and cranial nerve examination. Motor examination revealed bilateral weakness and atrophy of the small hand muscles in his hands (MRC grade 4/5) and significant weakness and atrophy of the distal leg musculature (foot dorsiflexion, eversion, and inversion were MRC grade 0/5). Muscle stretch reflexes in the upper extremities were intact, however they could not be elicited at the patellae or ankles and the plantar responses were flexor bilaterally. Sensory examination disclosed mild sensory loss of pinprick and light touch in a glove and stocking distribution. Proprioception and vibration sense were intact testing in his hands and feet. Tests of cerebellar function revealed no abnormalities and examination of his gait showed bilateral foot drop.
Family history
A detailed family history was obtained from at least two family members to corroborate whether other relatives were affected or unaffected. The parents of the patient have no clinical history to suggest that they were affected (Fig. 1). Furthermore, there is no history to suggest that any other member of the extended family was affected including paternal or maternal grandparents, uncles, aunts or cousins.
The index patient (III-5) had four siblings and of these, three are still alive. An eighty year old brother (III-4) was asymptomatic until age 40 years when he started having difficulty with his gait. Over the ensuing 40 years, his symptoms progressed and currently he is wheel chair bound. A neurological examination performed in India confirmed the presence of a severe neuropathy. This individual has three sons, 45 to 52 years of age who by history are asymptomatic and who declined to participate in the study. Another brother (III-3) died in his early thirties and did not have any neurological complaints. There are two sisters one (III-7) age 65 years, is normal and the other (III-8) age 60 years has leg weakness and a “funny walk”. Based upon her history, it is likely she is affected. Two of his cousins III-1 and III- 2 agreed to participate in the study and are asymptomatic. The index patient has three sons whose ages range between 34 to 45 years and who are all asymptomatic. We have examined individual IV-1 age 40 years and both his neurological exam and an EMG study were normal. His brother, IV-2, age 34 years has a normal neurological examination. The third son age 45 years is asymptomatic and declined to participate in the study.
Laboratory testing
The following investigations were normal or negative: Complete blood count (CBC) and differential, routine serum chemistries, comprehensive metabolic panel, Vitamin B12, folate, methylmalonic acid, serum protein electrophoresis and immunofixation, erythrocyte sedimentation rate, serological testing for rheumatoid factor, anti nuclear antigen, acetylcholine receptor antibody and Lyme disease.
Magnetic resonance scans of the brain and lumbosacral spine were within normal limits.
A number of electrophysiological studies have been performed over the years. The most comprehensive was performed recently and is summarized in Table 2.
Click to view | Table 2. Case 2, Patient 111-5-Nerve Conduction Study |
It demonstrates the presence of a neuropathy affecting the motor more than the sensory nerves. For example, although the evoked motor responses could not be elicited in the tibial nerves, the sural nerve potentials were still obtained. Furthermore, there were demyelinating features studying both ulnar and median nerves with evidence of conduction block, temporal dispersion and prolonged F-wave latencies.
Genetic testing
The following genetic tests were performed by Athena Diagnostics Inc. (Worcester, Massachusetts, USA) and were negative: tests for spinal and bulbar muscular atrophy, duplication/deletion analysis of PMP22, exon sequencing analysis of the Cx32, MPZ, PMP22, EGR2, NFL andGDAP1.
However, sequence analysis of the exon 7B3 of PRX gene identified a 15 bp deletion in the nucleotide position 1900-1914 translating to codon 634-638 in periaxin protein.
We developed a polymerase chain reaction (PCR) based technique to detect this 15 bp deletion mutation. PCR was carried out in a final volume of 50 µL with 40 ng of genomic DNA, 1X Taq buffer (50 mM KCl, 10 mM Tris-HCl,1.5 mM MgCl2 and 0.1% Triton X-100), 2.5 units Taq DNA polymerase (Promega), 0.4 mM dNTP (Applied Biosystems) and 0.5µM of each primer (Forward: 5’-GATGTGCACCTCCCAGAAGT -3’ and Reverse: 5’-CAGCCATCTCAGGATTTTA-3’). This was subjected to PCR under the following cycling parameters: 94 °C 2 min, 30 cycles (94 °Cfor 60 s, 55 °C for 60 s, 70 °C for 60 s) with a final extension at 72 °C for 14 min. Fifteen µL of the reaction product was then resolved by gel electrophoresis (6% polyacrylamide gel at 136 mV for 60 min BIORAD). The gel was stained (SYBR gold) and imaged for further analysis. Wild type DNA results in PCR product of 182 bp while the mutant DNA can be detected by visual analysis of a 167 bp fragment (Fig. 2A). These PCR products were confirmed by direct sequencing (Fig. 2B).
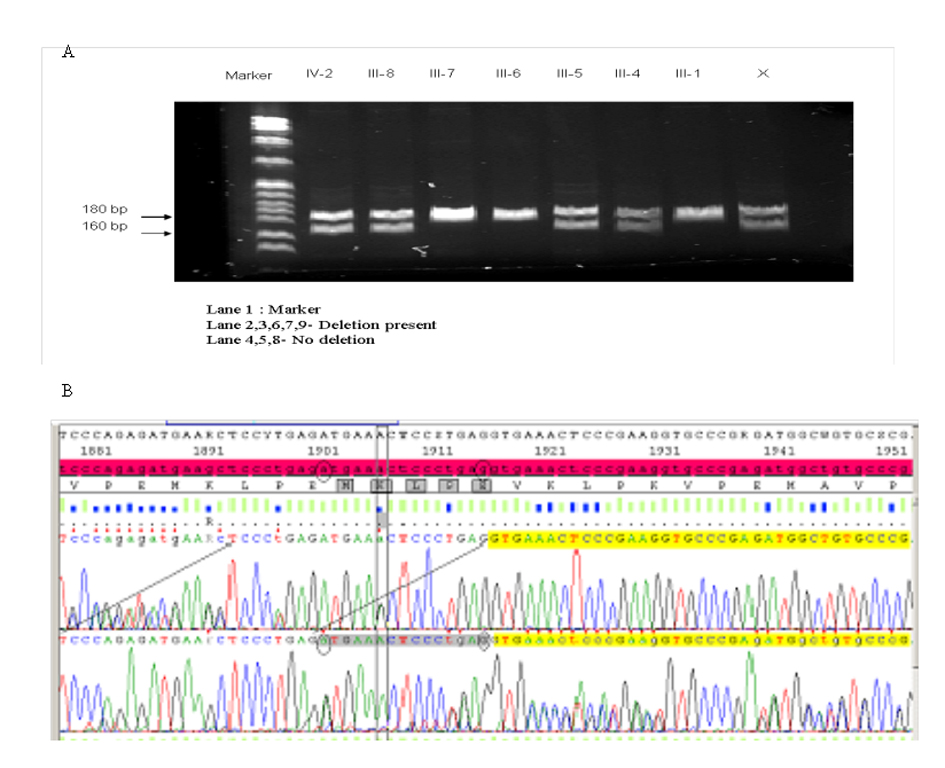 Click for large image | Figure 2. Gel showing the results of (A) the results of PCR for the 15 bp deletion mutation in periaxin gene in the family of the index patient (B) the results of the sequencing of the PCR products. |
Following IRB policies and procedures, blood samples were obtained and DNA extracted by standard methods from all available family members and unrelated control individuals. Employing our PCR based technique, the DNA was subjected to further analysis.
In this family the 15 bp deletion was detected in the patient’s affected brother (III-4), one of his sisters (III-8) who is probably affected and an asymptomatic son (IV-2) (Fig. 2A).
To measure the frequency of this mutation, we genotyped one hundred unrelated individuals from a variety of ethnic groups (30 Asian Indians, 10 African-Americans and 30 Caucasians). Interestingly, this deletion mutation was detected in one Asian individual who also emigrated from Gujarat, India, the same state as the index patient. This 45 year old man clinically had evidence of a mild neuropathy but had been diabetic for more than five years and this may be a contributing factor in the development of the neuropathy. He was not available for EMG testing and no further information is known regarding family history.
To investigate the epidemiology of this mutation further, we screened 100 unrelated immigrants from Gujarat, India but did not identify anyone else carrying this mutation.
Athena diagnostics database
A database is maintained by Athena Diagnostics, Inc. (Worcester, Massachusetts, USA). In this database, there is one other individual carrying the 15 bp deletion mutation in the PRX gene of about 15,000 DNA samples analyzed representing a frequency of 0.006%. The 78 bp mutation deletion is more frequent and has been observed in 32 individuals and has a frequency of 0.21%. Although there is no demographic or detailed clinical data available on any of these patients in the database, they are not representative of the normal population. It is likely that the majority of DNA samples in this database are from patients suffering a neuropathy as opposed to myopathies or other neuromuscular conditions.
| Discussion | ▴Top |
Clinically, patients with CMT4F typically present with severe leg weakness by age 10 years and with involvement of the hands by age 15 years. The neuropathy is further characterized by with slow or little progression. Electrophysiologically, there are demyelinating features and the motor and sensory nerve conduction velocities are severely reduced with values less than 20 m/sec.
The neuropathy that developed in the both of these patients is, by both clinical and electrophysiological criteria, characterized by greater involvement of the motor than sensory fibers. There are also demyelinating features and overall, the pattern in both patients suggest multi-focal motor neuropathy. In contrast to the reported patients with CMT 4F, the neuropathy in these patients is clinically and electrophysiologically less severe with a later age of onset.
Mutations in the human PRX gene have been shown to result in AR Dejerine-Sottas Syndrome (DSS) and AR CMT4F. In a recent report, Marchesi et al [17] discussed four cases of ARCMT caused by novel mutations in the PRX gene and reviewed mutational analysis of twenty-three previously reported patients. All the mutations reported are located in the exon 7 of the PRX gene and cause truncation of the protein with loss of the C-terminus indicating an important role for this domain in periaxin function. The primary structure of periaxin is comprised of a series of penta- and higher order repeats extending from amino acid 430-730 that separates a strongly basic N-terminal from the acidic C-terminal segment of the protein. This repeat-rich region is less flexible and serves to extend the protein and keep the acidic and basic domains well separated [6]. No mutations or alterations involving this repeat region have been previously reported.
Interestingly, the deletion mutations in these two patients also involve exon 7. The 15 bp mutation (codon 634 to 638) results in deletion of the two adjacent penta-repeats and the 78 bp mutation (codon 495 to 520) results in deletion of three pentamers and two tripeptide spacers (Fig. 3). Both these deletion mutations affect the core repeats and disrupt higher order repeats in the central repeat region. However they do not affect the mRNA reading frame and unlike the previously described PRX mutations, do not result in premature truncation of the periaxin protein.
 Click for large image | Figure 3. Showing the repeat region comprising of amino acids 430 to 730, deletion in this region leads to disruption of the core penta and tri repeats and also affects the higher order repeats in the region. The amino acids deleted are indicated in bold Italics. The 15 bp deletion in Case 1, index patient III-5 results in deletion of five amino acids causing disruption of two adjacent penta-repeats. The 78 bp deletion in Case 2 results in deletion of twenty-six amino acids leading to disruption of two penta-repeats and deletion of three pentamers and two spacer tri-peptide repeats. |
It is possible that these deletion mutations have occurred by chance in our patients and represent the random association of rare genetic changes with a neuropathy. However, there is a possible mechanism in which these deletions could potentially be disease producing.
All of the reported mutations causing DSS or CMT4F identified in the PRXgene follow an AR pattern of transmission. In both patients, none of the parents are affected suggesting that although at least one parent in each family carries the mutation, they remain unaffected. In the family of case 2, the 15 bp deletion mutation is associated with late onset neuropathy in two definitely affected members (III-4 and III-5) and one probably affected (III-8). Although we do not have electrophysiologic testing to characterize the neuropathy in these individuals, the presence of multiple affected members in this family does suggest a genetic cause. In both families, none of the children of affected individuals who are at risk are symptomatic and have no clinical features to suggest a neuropathy. Overall, in both families, this pattern is consistent with AR transmission and suggests that a mutation in the second allele is present although this was not confirmed by comprehensive exonic sequencing. However, intronic changes are not detected by exonic sequencing and it is biologically plausible that such a change could affect gene expression. In that case, the disease would maintain an AR pattern of transmission. This hypothesis could be explored further by complete genomic sequencing of the PRX gene and protein expression analysis in peripheral nerve tissues. We approached both patients about performing a nerve biopsy to investigate periaxin protein expression but they declined.
From an epidemiological point of view, it is interesting to note that another unrelated individual from Gujarat carries the same deletion. Pooling the index patient with unrelated individuals from Gujarat show a deletion mutation frequency of 2/130 (1.54%) for our lab compared with about 1/15,000 (0.013%) in the Athena database.
This is the first report of deletions in the repeat region of the PRX gene.Here we report the association of deletion mutations in the repeat region of the PRX gene with a predominantly motor neuropathy in two unrelated patients. Whether or not this is a chance association of an uncommon form of neuropathy with a rare genetic change is not clear. Further investigations of more patients and families carrying these deletion mutations will be needed to confirm this association.
Acknowledgments
We acknowledge the support of the Neurogenetics Foundation.
Conflicts of Interest
None declared.
| References | ▴Top |
- Skre H. Genetic and clinical aspects of Charcot-Marie-Tooth's disease. Clin Genet. 1974;6(2):98-118.
doi pubmed - Dyck PJ, Lambert EH. Lower motor and primary sensory neuron diseases with peroneal muscular atrophy. II. Neurologic, genetic, and electrophysiologic findings in various neuronal degenerations. Arch Neurol. 1968;18(6):619-625.
doi pubmed - Harding AE, Thomas PK. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain. 1980;103(2):259-280.
doi - Bernard R, De Sandre-Giovannoli A, Delague V, Levy N. Molecular genetics of autosomal-recessive axonal Charcot-Marie-Tooth neuropathies. Neuromolecular Med. 2006;8(1-2):87-106.
doi - Auer-Grumbach M, Fischer C, Papic L, John E, Plecko B, Bittner RE, Bernert G, et al. Two novel mutations in the GDAP1 and PRX genes in early onset Charcot-Marie-Tooth syndrome. Neuropediatrics. 2008;39(1):33-38.
doi pubmed - Gillespie CS, Sherman DL, Blair GE, Brophy PJ. Periaxin, a novel protein of myelinating Schwann cells with a possible role in axonal ensheathment. Neuron. 1994;12(3):497-508.
doi - Menard HW. The East Pacific Rise. Science. 1960;132(3441):1737-1746.
doi pubmed - Dytrych L, Sherman DL, Gillespie CS, Brophy PJ. Two PDZ domain proteins encoded by the murine periaxin gene are the result of alternative intron retention and are differentially targeted in Schwann cells. J Biol Chem. 1998;273(10):5794-5800.
doi pubmed - Sherman DL, Fabrizi C, Gillespie CS, Brophy PJ. Specific disruption of a schwann cell dystrophin-related protein complex in a demyelinating neuropathy. Neuron. 2001;30(3):677-687.
doi - Gillespie CS, Sherman DL, Fleetwood-Walker SM, Cottrell DF, Tait S, Garry EM, Wallace VC, et al. Peripheral demyelination and neuropathic pain behavior in periaxin-deficient mice. Neuron. 2000;26(2):523-531.
doi - Boerkoel CF, Takashima H, Stankiewicz P, Garcia CA, Leber SM, Rhee-Morris L, Lupski JR. Periaxin mutations cause recessive Dejerine-Sottas neuropathy. Am J Hum Genet. 2001;68(2):325-333.
doi pubmed - Guilbot A, Williams A, Ravise N, Verny C, Brice A, Sherman DL, Brophy PJ, et al. A mutation in periaxin is responsible for CMT4F, an autosomal recessive form of Charcot-Marie-Tooth disease. Hum Mol Genet. 2001;10(4):415-421.
doi pubmed - Takashima H, Boerkoel CF, De Jonghe P, Ceuterick C, Martin JJ, Voit T, Schroder JM, et al. Periaxin mutations cause a broad spectrum of demyelinating neuropathies. Ann Neurol. 2002;51(6):709-715.
doi pubmed - Kijima K, Numakura C, Shirahata E, Sawaishi Y, Shimohata M, Igarashi S, Tanaka T, et al. Periaxin mutation causes early-onset but slow-progressive Charcot-Marie-Tooth disease. J Hum Genet. 2004;49(7):376-379.
doi pubmed - Otagiri T, Sugai K, Kijima K, Arai H, Sawaishi Y, Shimohata M, Hayasaka K. Periaxin mutation in Japanese patients with Charcot-Marie-Tooth disease. J Hum Genet. 2006;51(7):625-628.
doi pubmed - Kabzinska D, Drac H, Sherman DL, Kostera-Pruszczyk A, Brophy PJ, Kochanski A, Hausmanowa-Petrusewicz I. Charcot-Marie-Tooth type 4F disease caused by S399fsx410 mutation in the PRX gene. Neurology. 2006;66(5):745-747.
doi pubmed - Marchesi C, Milani M, Morbin M, Cesani M, Lauria G, Scaioli V, Piccolo G, et al. Four novel cases of periaxin-related neuropathy and review of the literature. Neurology. 2010;75(20):1830-1838.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Neurology Research is published by Elmer Press Inc.