

| Journal of Neurology Research, ISSN 1923-2845 print, 1923-2853 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Neurol Res and Elmer Press Inc |
| Journal website https://www.neurores.org |
Review
Volume 12, Number 3, October 2022, pages 93-113

Review of Huntington’s Disease: From Basics to Advances in Diagnosis and Treatment
Natalia Gonzalez Rojasa, Martin Emiliano Cesarinia, Guillermo Pekera, Gustavo Andres Da Prata, Jose Luis Etcheverrya, Emilia Mabel Gattoa, b, c
aInstituto Neurociencias Buenos Aires (INEBA), Guardia Vieja 4435, CABA, Argentina
bSanatorio de la Trinidad Mitre, Bartolome Mitre 2553, C1039 CABA, Buenos Aires, Argentina
cCorresponding Author: Emilia Mabel Gatto, Instituto de Neurociencias Buenos Aires, Area de Enfermedad de Parkinson y Movimientos Anormales, Guardia Vieja 4435, CABA, Buenos Aires, Argentina
Manuscript submitted March 14, 2022, accepted June 30, 2022, published online October 22, 2022
Short title: Review of Huntington’s Disease
doi: https://doi.org/10.14740/jnr721
- Abstract
- Introduction
- Methods
- Epidemiology
- Genetics
- Pathogenesis
- Clinical Presentation
- Differential Diagnosis
- Biomarkers
- Treatments
- Conclusions
- References
| Abstract | ▴Top |
We conducted the present review facing the enormous growth of scientific knowledge in Huntington’s disease (HD) and the need for a practical update for general neurologists. HD is a devastating neurodegenerative disease of autosomal dominant inheritance and full penetrance, caused by an expansion of the cytosine-adenine-guanine (CAG) trinucleotide in the huntingtin gene located on chromosome 4. The clinical phenotype varies according to the age of presentation, but it is mainly characterized by cognitive, motor and psychiatric disturbances. Many mechanisms were raised trying to explain the path to neurodegeneration, including disruption of proteostasis, transcription and mitochondrial dysfunction as well as direct toxicity. There has been tremendous progress regarding disease pathogenesis, clinical management and promising new therapeutic avenues including disease-modifying treatments that pose a challenge and a need for a practical approach to be taken by movement disorders specialists and general neurologists.
Keywords: Huntington’s disease; Genetics; Mechanisms; Diagnosis; Treatment
| Introduction | ▴Top |
Huntington’s disease (HD) is an autosomal dominant, neurodegenerative disorder with complete penetrance caused by a cytosine-adenine-guanine (CAG) trinucleotide repeat expansion in the huntingtin (HTT) gene (previously called IT-15) on chromosome 4. The expanded CAG results in a mutant protein (huntingtin (HTT)) rich in glutamine amino acids (polyQ), with toxic properties to the cell. It was first described by an American physician, Charles Waters in 1841, but it was not until 1872 when George Huntington wrote a detailed report of hereditary chorea and coined the name by which we know it today. In 1983, a connection to chromosome 4 was recognized and 10 years later the causative gene was identified in the population of Maracaibo, Venezuela [1]. Although there are many symptomatic treatment options, several ongoing studies may be the long-awaited response to this devastating disease.
| Methods | ▴Top |
We conducted a research of the literature through PubMed on HD. Keywords such as Huntington’s disease, pathogenesis, genetics, clinical variants and differential diagnoses were used. We also researched information regarding treatments, considering both the traditional ones as well as more recent advances in this area. The aim was to summarize all these data and facilitate access to the information for both general neurologists and movement disorders specialists.
| Epidemiology | ▴Top |
The worldwide prevalence is extensively varied with an equal distribution between men and women. It is estimated in 5-10 cases per 100,000 inhabitants, with lower numbers in East Asians, and higher in white Europeans. Annual incidence varies between one and four cases per million inhabitants. The advent of the genetic testing allowed locating the origin of the disease in Western Europe (France, Germany and the Netherlands), with subsequent expansion towards America, England, South Africa and Australia.
Isolated populations with a very high prevalence were reported in Maracaibo (Venezuela) and Canete (Peru) in South America, in Tasmania in Oceania (17.4 per 100,000 people), and in a small region of Scotland called Moray Firth [2] (Fig. 1).
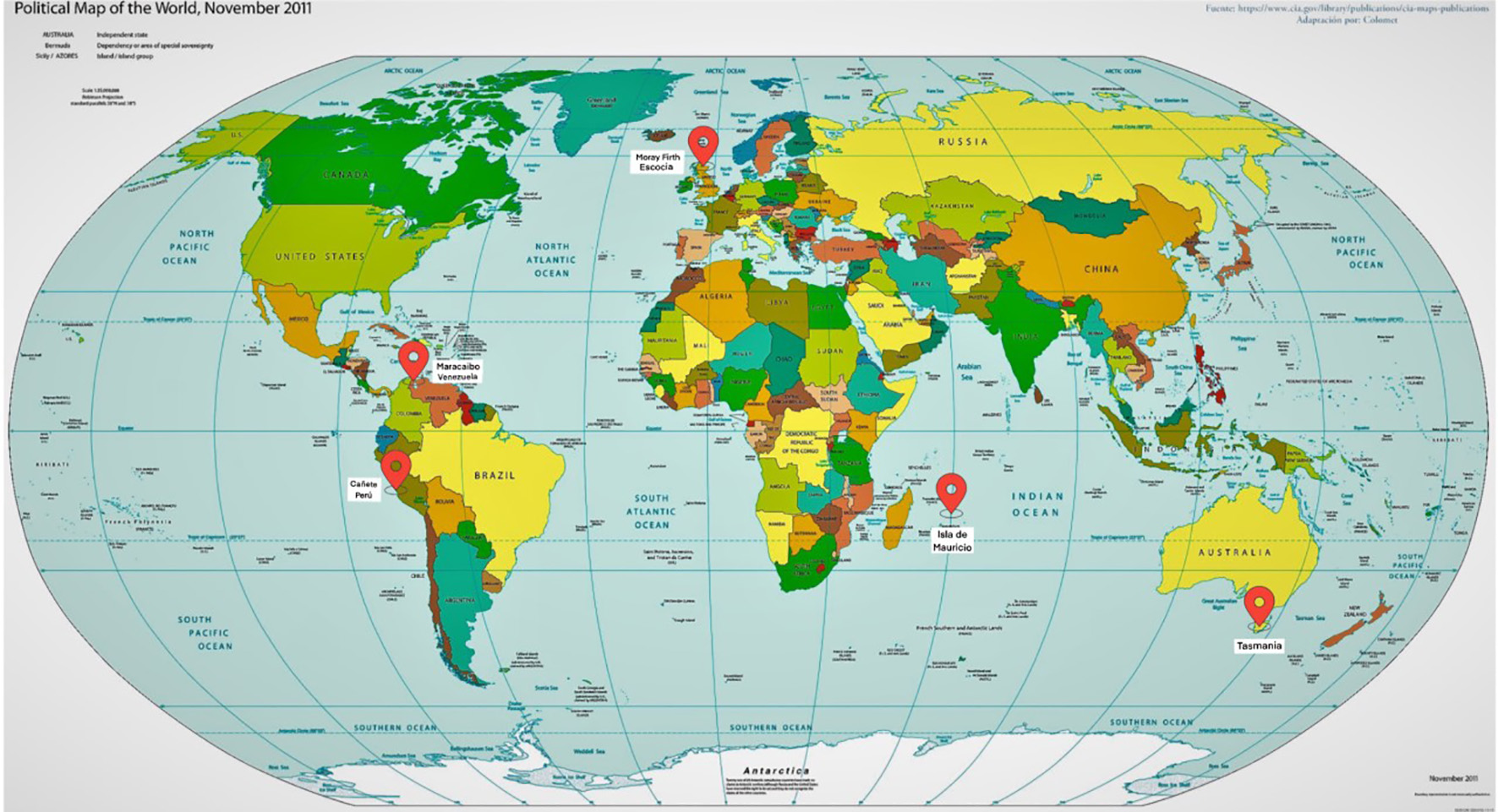 Click for large image | Figure 1. Cluster worldwide populations with the highest prevalence. |
The Peruvian cluster was first described in 1990, by identifying 30 cases among 392 individuals corresponding to a single family, in the Canete Valley. The first case reported within this family group could date from 120 years. Both filial data and personal traits all corresponded to mixed race people; therefore, the HD mutation is believed to be originated from European immigrants with subsequent local mixing. This small population was considered the starting point from where HD then spread to the entire country [3] (Fig. 2).
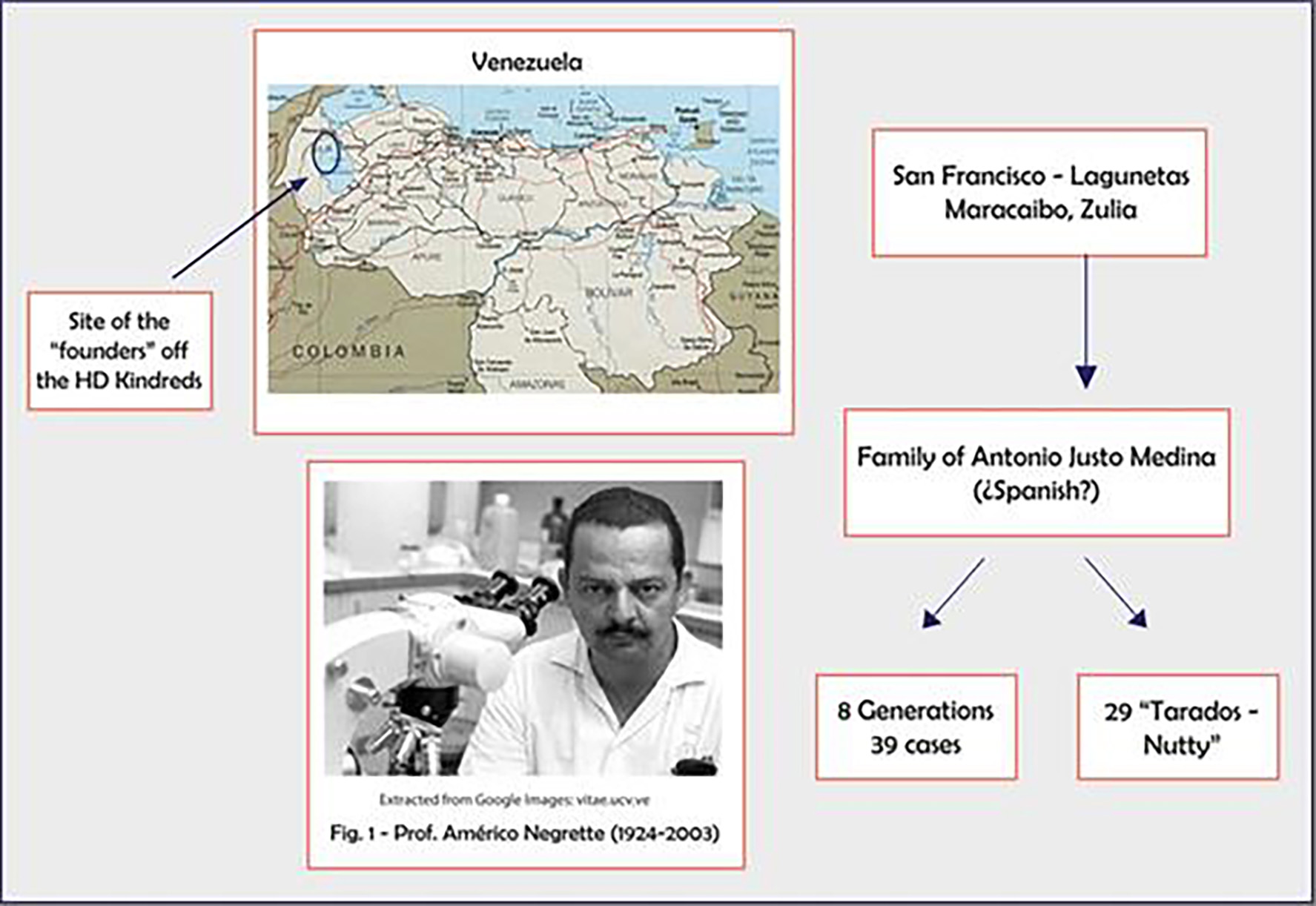 Click for large image | Figure 2. Prof. Negrete, Americo, who identified the Zulia cluster at Maracaibo, Venezuela. |
In North America, the highest prevalence was reported by Fisher and Hayden [4] among white people in Canada (17.27 per 100,000). In the United States, the age-adjusted cumulative incidence rate was 1.22/100,000 persons, and age-adjusted diagnostic frequency is 6.52/100,000 persons.
On the other hand, studies from Asia show ranges from 0.11 per 100,000 to 0.72 per 100,000 markedly lower than those in most of Oceania, Western Europe and the United States [5].
Results from Europe have varied with the years, as seen comparing studies in 1989, where the prevalence found was 4.47/100,000 population to 2010 with 5.16/100,000 [2].
| Genetics | ▴Top |
The HTT gene, located in chromosome 4p16.3, was first identified in 1993 [6]. It encodes for the huntingtin (HTT) protein. Though the precise function of the protein remains unknown, it apparently plays an important role in neurons and is essential for normal embryogenesis. Huntingtin is found in many tissues, with the highest levels of activity in the brain. Within cells, this scaffold protein may be involved in chemical signalling, gene transcription, vesicular transport, protein binding and protection from apoptosis [7, 8]. Huntingtin exon 1 sequence contains 17 amino acids at the N-terminus (Fig. 3), a naturally polymorphic CAG trinucleotide repeat, and a 51-residue proline-rich domain (PRD). The CAG repeats are translated into an expanded polyglutamine tract. Accordingly, HD is one of the polyglutamine repeat expansion diseases which also include some autosomal dominant spinocerebellar ataxias and spinal and bulbar muscular atrophy, considered thus far a monogenic disease [9].
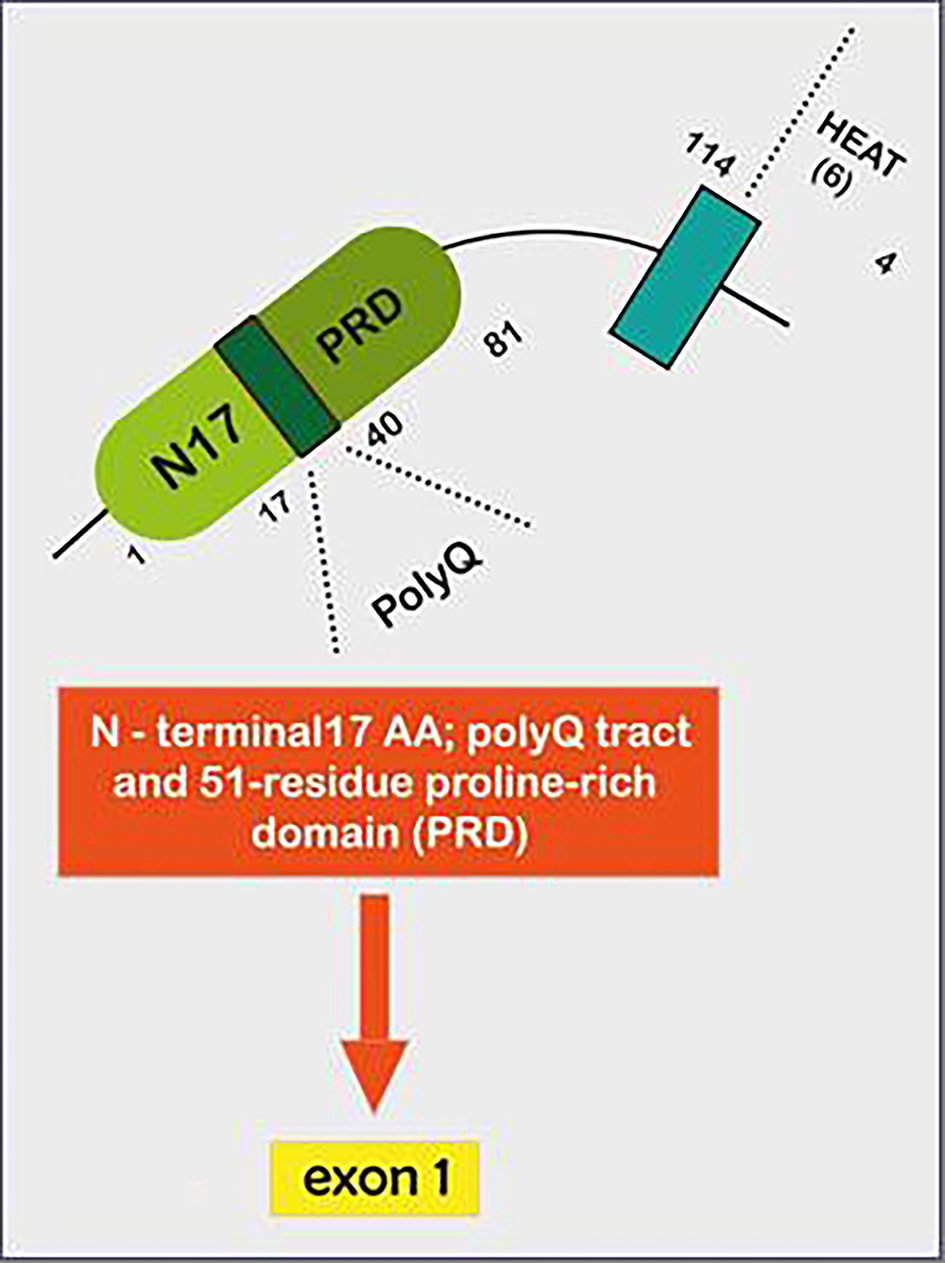 Click for large image | Figure 3. Illustrative figure showing how Huntingtin exon 1- represents a particular DNA segment known as a CAG trinucleotide repeat. This fragment is made up of a series of three DNA nitrogenous bases (cytosine, adenine and guanine) that appears multiple times in a row. The normal CAG repetitions are 10 to 35. HD is caused by expansion of that CAG trinucleotide that encodes the 17 AA N-terminal 17; a polyQ tract and 51-residue proline-rich domain (PRD), which is translated into a corresponding polyglutamine tract that turns medium spiny neurons in the striatum exceptionally susceptible to apoptosis, leading to their dysfunction and early death. HD: Huntington’s disease; CAG: cytosine-adenine-guanine. |
The range from 17 to 20 repeats is the most commonly found in unaffected individuals. Repeats between 27 and 35 (intermediate alleles) are infrequent and not associated with disease manifestations; however, they are meiotically unstable (mosaicism) and can expand over the generations into the pathological range, as was seen in paternal transmission. The same premise applies for cases of incomplete penetrance (36 - 39 repetitions), where the repetition range is clearly associated with positive symptoms, but considering a late onset age of presentation, some individuals may never manifest symptoms, but transmit the mutation to future generations without any previous history [10]. The vast majority of adult-onset cases have 40 - 50 CAGs, whereas expansions of 50 and more are associated with juvenile onset (Westphal variant). There is an inverse correlation between the age of onset and the number of CAG repeats, thus, longer repeats are associated with an earlier presentation. Despite that, the extension of the CAG triplet accounts only for 70% of this variance. Meanwhile, the remaining percentage is represented by other modifying factors. The largest genome wide association study (GWAS) in HD identified a number of genes involved in DNA repair that might enhance mosaicism and promote the expansion, impacting the age of motor onset. The most important ones are located on chromosomes 8 and 15, in charge of onset-hastening and onset-delaying, respectively [11-13] (Table 1).
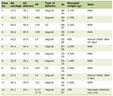 Click to view | Table 1. Genetic Modifiers of Blood DNA Somatic Expansion Scores of the HTT CAG Repeat |
Many reports established that pure CAG length, rather than the final number of encoded-glutamine, is the key of HD pathophysiology. Motor age at onset is then partly predicted by the inherited number of CAG repeats along with other modifying genes and epigenetics. Some other DNA repair genes, such as FAN1, MLH 1, MLH3, MSH3 and RRM2B/UBR5, might affect disease severity by modifying somatic expansion of the CAG repeat, through the entire patient’s life [14, 15].
| Pathogenesis | ▴Top |
The molecular alterations are not yet fully understood. Despite toxicity results from a gain of function of the mutant protein, the wild type loss of function also contributes to the pathophysiology of the disease. It plays an important role in brain development, so its inactivation cannot be entirely dismissed as a road to neurodegeneration, as deletion of the huntingtin gene prior to neural development showed to be lethal in mice models.
Therefore, HD has been suggested as a neurodevelopmental disorder characterized by abnormalities in the cortex forming process as well as in mitosis and cell cycle progression [16].
A mechanism recently reported could change the paradigm regarding the number of polyglutamines and the age of onset of the disease. CAA is considered an interruption triplet that alternates with the CAG along the gene, and even though it does not interfere with HD pathogenesis as it codes for glutamine itself too, the lack of CAA interruption may increase the polyglutamine expansion instability posing a higher risk of transmission to the offspring. This might be particularly relevant for people with repeat lengths close to the pathological threshold.
There are descriptions about rare individuals with two copies of the mutant allele. Patients with biallelic CAG or compound heterozygous expansion have been described in diverse ethnic groups and the prevalence ranges from 0.1% to 0.4%. Clinical features are similar to heterozygous individuals regarding age of onset and disease course, although some reports suggested a more severe clinical course with a more rapid progression in the biallelic group. Possible explanations for biallelic HD include non-paternity, an intermediate or reduced penetrance HTT allele in the unaffected parent, a full mutation allele in an asymptomatic parent who died prematurely or HTT uniparental isodisomy [17].
Mutant huntingtin aggregation
Polyglutamine aggregates in the brain were initially found in the nucleus and subsequently in the cytoplasm of neurons of HD patients. Besides the main component is still the expanded mutant huntingtin, many other proteins, including ubiquitin, proteasome subunits and chaperones, transcription factors, and the wild-type huntingtin were also found.
Studies showed that the aggregates in adult-onset HD are typically cytoplasmic, while younger patients were more frequently located in the nucleus. These perinuclear aggregates proposed to be neurotoxic, causing cell death by abnormal activation of the cell cycle leading to apoptosis [18]. In HD, oligomers of HTT protein sequestered chaperones, proteosome subunits, transcription factors and wild-type huntingtin (wHTT), weakening the protective capacity of the cell inducing a higher autophagy flux rate [19].
In addition to the aforementioned regarding HTT fragments, the potential importance of smaller N-terminal fragments is highlighted by their presence in HD post-mortem brains and by the fact that nuclear inclusions are only detected by antibodies to the N-terminus of HTT. The precise length of these fragments and the underlying mechanism remains unknown. Studies by Sathasivam et al showed that CAG repeat length dependent aberrant splicing of exon 1 HTT results in a short polyadenylated mRNA, which is latter translated into an exon 1 HTT protein. Given the last has shown to be highly pathogenic in HD mouse models, the aberrant splicing of HTT mRNA provides a mechanistic basis for the molecular pathogenesis of HD and encourages the search for possible RNA-targeted therapeutic strategies [20].
Huntingtin toxic fragments
A key molecular feature is the cleavage of huntingtin in the cytoplasm followed by the translocation of the fragments into the nucleus of striatal neurons. The accumulation of these fragments comes from proteolysis by caspases, calpains and other proteases. Although both types, wild and expanded, get cleaved, the presence of mutant fragments increases toxicity and has been proposed as the first step in the process of neurodegeneration [21].
Disruption of transcription
Several studies have demonstrated that expression profiles of a vast number of genes are deeply modified, mainly those related to neurotransmitter receptors and ion channels and brain-derived neurotrophic factor (BDNF), a pro-survival factor produced at the cortex which promotes striatal neurons survival.
The transcriptional activation and suppression controlled by chromatin acetylation has been found to be levelled down in HD pathology. Two enzymes with opposite functions are in charge of the acetylation and deacetylation of histone proteins, histone acetyl-transferases and histone deacetylases (HDACs), favoring chromatin structure modifications (euchromatin (non-compact form or active state) or heterochromatin (compact or inactive form which inhibits transcription and replication)), respectively. The imbalance between these processes contributes to an altered transcriptional programme.
Mutant huntingtin interacts with many transcription regulator factors, involving: 1) cell proliferation, DNA damaged repair (p53, CREB, CBP, MSK-1); 2) energy metabolism (PGC-1a); 3) organelle, vesicle transport and factors that regulate the dynactin/dynein activity; 4) genes related with dopamine 2 receptors (D2R) [22] (Fig. 4).
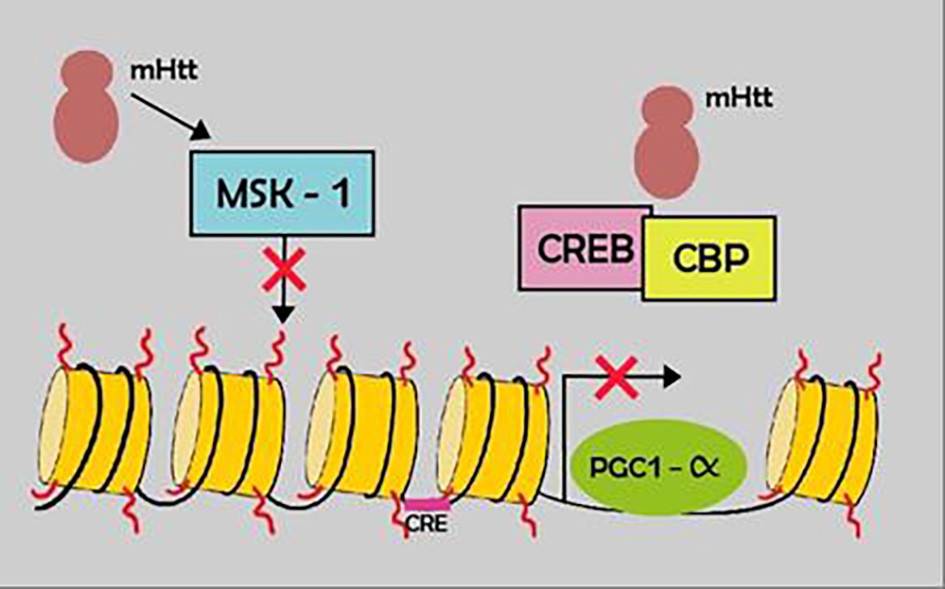 Click for large image | Figure 4. Example of the transcription impairment in HD. Mutant htt results in down regulation of certain transcription factors (MSK-1) and sequestration of others, CREB and CBP. HD: Huntington’s disease. |
Alterations in gene expression beyond transcription: epigenetics and noncoding RNAs
In addition to the DNA methylation, HTT promotes the conformation of inactive, condensed heterochromatin. Sirtuins (SIRT) are a family of nicotinamide adenine dinucleotide (NAD)-dependent HDACs with multiple cellular functions which are normally blocked by whtt, while Htt induces its overexpression, affecting cell survival and mitochondrial biogenesis [23].
Gene expression is also influenced by non-coding RNAs. RNA toxicity mechanisms include aberrant protein-RNA interactions and protein sequestration, but also the hairpin secondary structure formed by CAG-RNA resembles the double-stranded RNA structures that are substrates for Dicer, dividing them into shorter repeats that silence specific genes [24] (Figs. 5, 6). All these pathways appear as a novel target therapy for HD that will be more extensively discussed later.
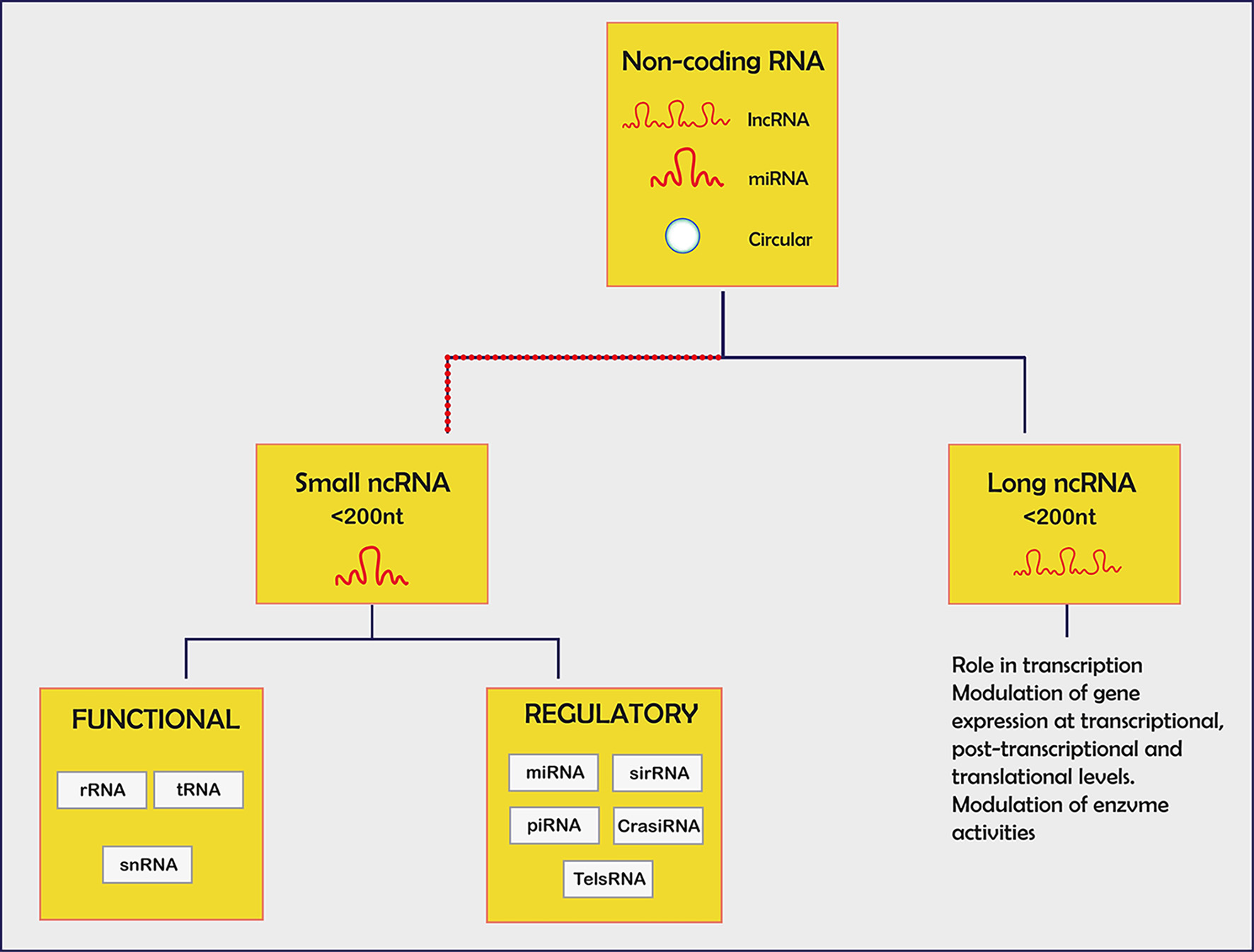 Click for large image | Figure 5. Non-coding small RNA with each specified function. miRNA: mRNA degradation/block translation; siRNA: mRNA degradation, chromatin condensation; piRNA: interaction piwi family of proteins; lncRNA: chromatin remodelling/transcriptional and post-regulation/precursors for siRNAs. |
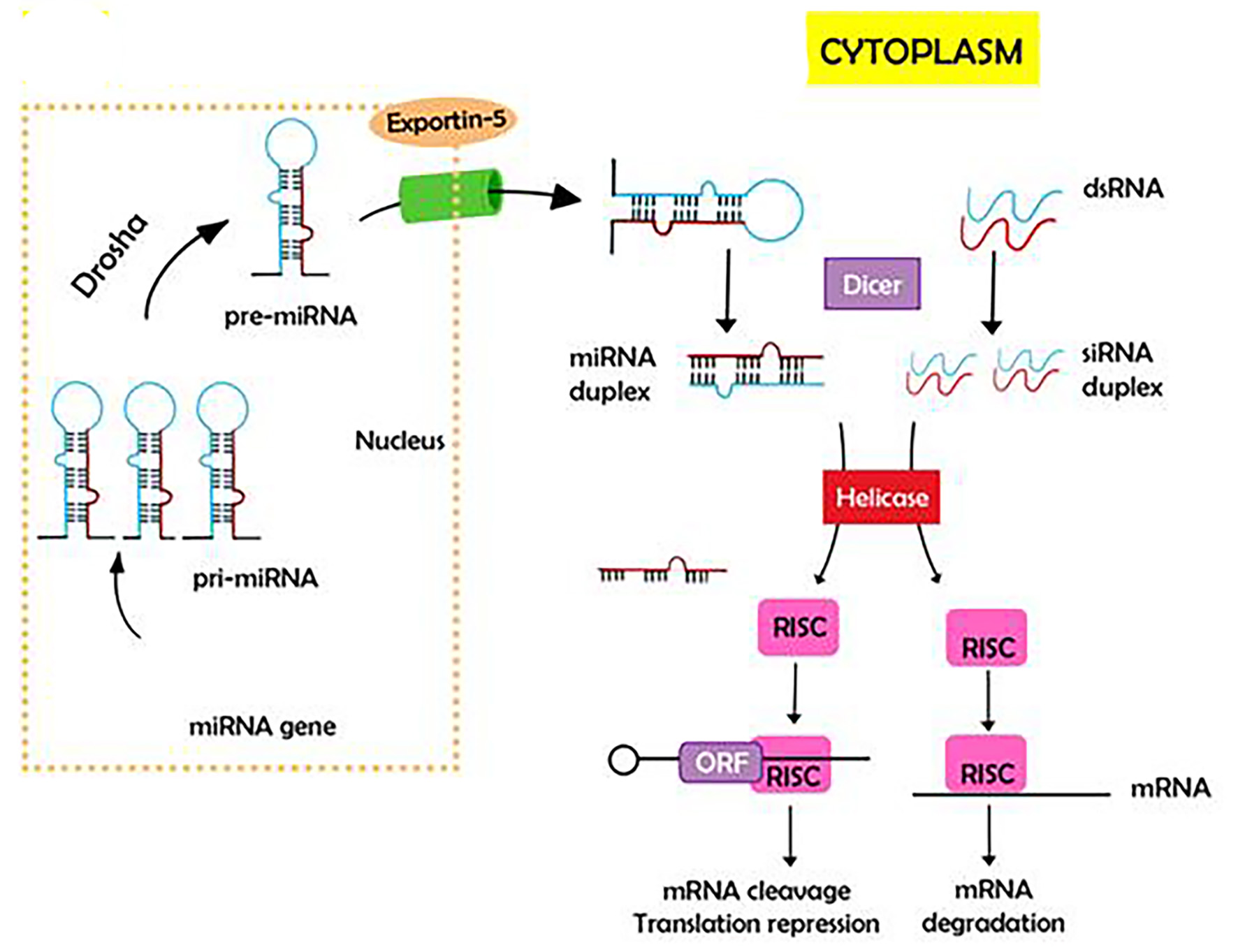 Click for large image | Figure 6. Gene expression is by non-coding RNAs. RNA toxicity mechanisms include aberrant protein-RNA interactions and protein sequestration, but also the hairpin secondary structure formed by CAG RNA resemble double-stranded RNA structures that are substrates for Dicer, dividing them into shorter repeats that silence specific genes. |
Altered synaptic plasticity and neuronal homeostasis
Synaptic abnormalities are premature pathological events in HD. HTT aggregates block axon transport, in addition to recruiting and annulling motor proteins, therefore inhibiting fast axonal transport. Loss of function of wHTT might be another point to consider, as in normal conditions, it enables vesicle transport.
The scaffold condition of the huntingtin protein relies on its capacity to interact, among other large number of proteins and factors, with huntingtin-associated protein 1 (HAP1), which in turn modulates other bindings with a microtubule protein complex (dynactin or kinesin), promoting a balance between anterograde and retrograde trafficking along the synaptic area [25].
Ubiquitin-proteasome system (UPS) and autophagy
The cell machinery possesses many different mechanisms to dispose of abnormal proteins and organelles; for instance, the UPS, and the autophagy-lysosome system [26].
Chaperones manage the initial step in the UPS, as they contribute to the correct protein folding, to be later ubiquitinized, and finally degraded inside the proteasome. The excess of unfolded proteins induces aggregation. Oligomers of HTT sequester chaperones, favoring protein accumulation [27].
Autophagy is a vesicular pathway through which abnormal proteins are included in a double membrane vesicle conforming an autophagosome that ultimately fuses to the lysosome promoting degradation. The failure of this system promotes cellular apoptosis by increasing misfolded inside the cytosol contributing also to increase the endoplasmic reticulum (ER) stress, oxidative stress, mitochondrial dysfunction among other apoptotic pathways [28].
Some reports describe a relation between the impairment in proteasome activity and the expression of polyglutamine-expanded huntingtin, either because some components of the UPS could be redirected into inclusions or some aggregates of HTT are resistant to the proteostasis network. Additionally, HD autophagosomes have a deficient axonal transport, impairing the correct autophagosome-lysosome fusion [26].
Mitochondrial dysfunction
HTT favors an increased entry of calcium inducing a failure at the respiratory chain. The final common pathway is a defective ATP production conducting the cell to apoptosis.
Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1a) is a nuclear coactivator that regulates the expression of certain genes that mediate mitochondrial biogenesis and respiration; in HD, its transcription is decreased. Additionally, HTT inhibits some membrane transport complex components, such as translocase inner membrane (TIM23), disturbing protein traffic through the mitochondrial membrane, leading to respiratory dysfunction and neuronal cell death [29].
All mentioned above determines an excessive production of reactive oxygen species (ROS) and free radicals, worsening the damage within the mitochondria, including the mytophagy pathway.
Cell-to-cell transmission of pathological aggregates
Although the exact mechanisms remain unknown, unconventional modes of secretion (exosomes) as well as tunneling nano-tubes have been proposed to contribute to migration of HTT aggregates from one neuron to another. The accumulation of polyglutamine proteins in HD serves as a starting point to increase the level of intracellular aggregation. Reports from Guo et al suggested a cell-to-cell transfer through actin-rich membrane bridges which connect cells and mediate the passage among neurons. This was supported by in vivo studies that endorse this hypothesis using human embryonic stem cell (hESC)-derived neurons. They proved that when these cells were unified with corticostriatal organotypic brain slices in mouse models, the former were capable of forming aggregates after 2 or 4 weeks supporting the seeding capacity of the HTT [30] (Fig. 7).
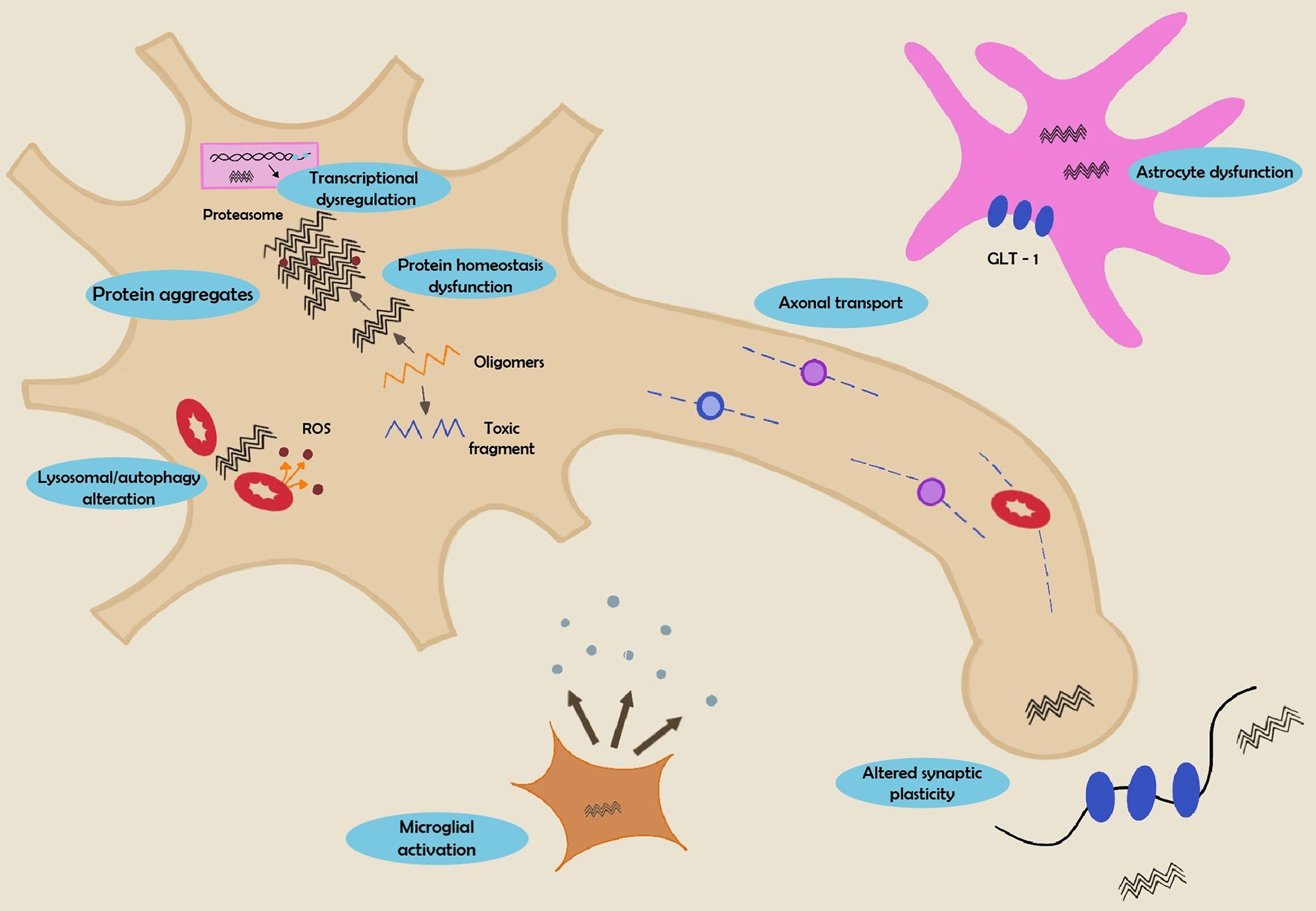 Click for large image | Figure 7. Summary of mechanisms involved in HD pathogenesis. HD: Huntington’s disease. |
Astrocyte and microglial dysfunction
Astrocytes provide a support network to neurons and prevent excitotoxicity by reuptaking extracellular glutamate. Pathological huntingtin aggregates are more pronounced in neurons than in glial cells, probably due to the absence of cell division in neurons or to an altered protein homeostasis system.
Experimental models support a correlation between GLT-1 glutamate transporter levels and HTT expression in astrocytes and a more severe phenotype when HTT was expressed not only in neurons but also in astrocytes. These findings support a main role of astroglia to the development of the disease [31].
Sphingosine receptors
Sphingosine phosphate 1 (S1P) is a potent signalling lipid, significantly reduced in HD. Its receptor, S1PR, is found on neurons, astrocytes and oligodendrocytes. The activation of both (SP1 and S1PR) promotes autophagy, reducing the aggregation of HTT through the activation of AKT and ErK survival pathways and BDNF production [32]. This fact could be taken into account in future treatments option (Fig. 8).
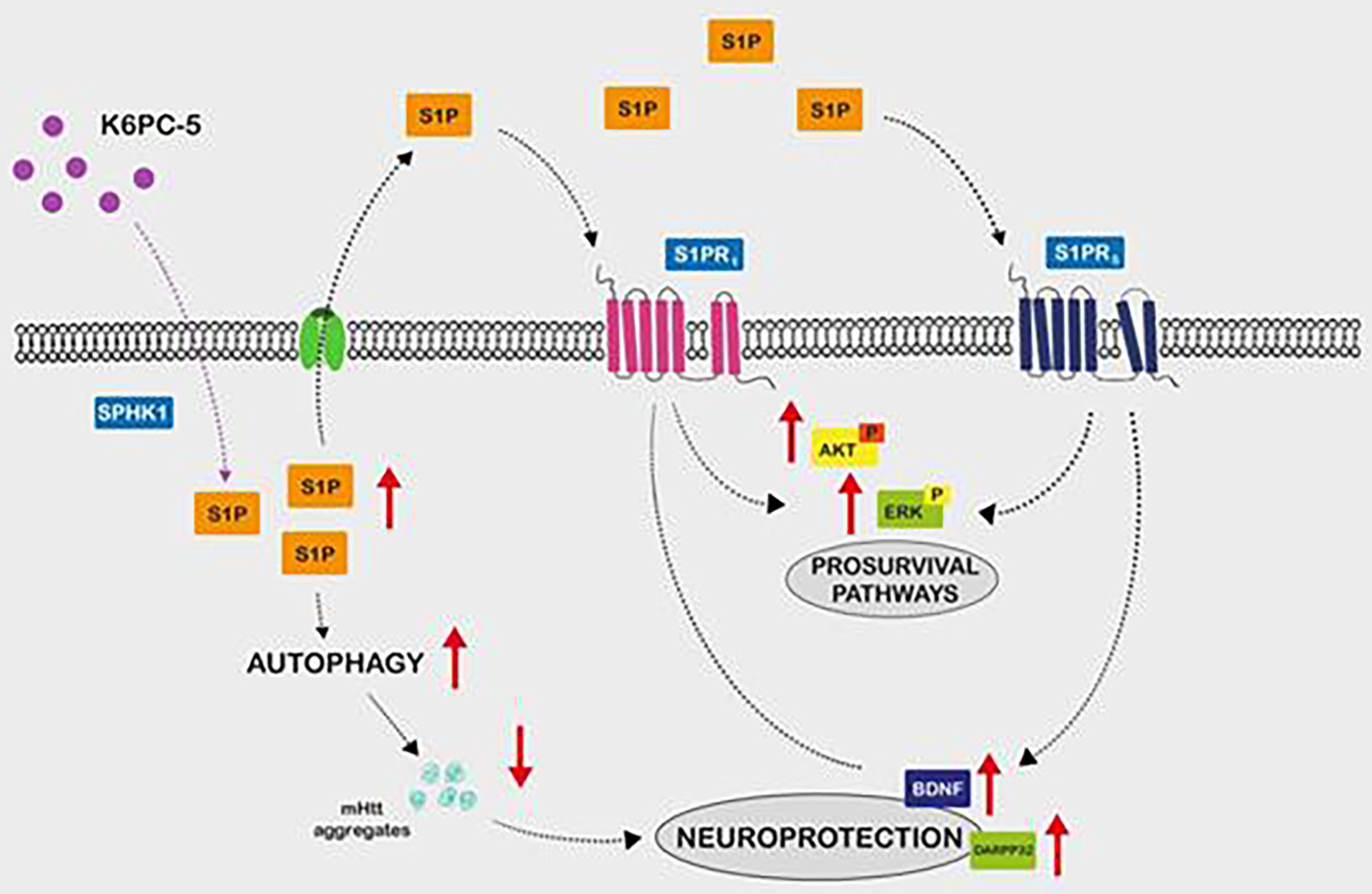 Click for large image | Figure 8. Sphingosine 1 phosphate is a potent signalling lipid. HD has been associated with a significant reduction of the sphingosine 1 phosphate (S1P) which action is mediated by the S1P receptors union. These receptors are located in neurons, astrocytes and oligodendrocytes. Experimental models identified autophagy stimulation, reduction of htt aggregation and activation of AKT and ErK pathways and BDNF production mediated by S1P signalling activation. Extracellularly level S1P stabilizes S1PR expression and activates pro-survival pathways. HD: Huntington’s disease. |
| Clinical Presentation | ▴Top |
HD is characterized by motor and non-motor manifestations, including cognitive decline and psychiatric disturbances. Mean age at disease onset is 45 years old, with duration of 17 - 20 years. Extrapyramidal motor signs and symptoms associated to HD define the clinical onset of the disease. However, cognitive and psychiatric aspects usually precede these by several years and should be taken into account [33] (Fig. 9).
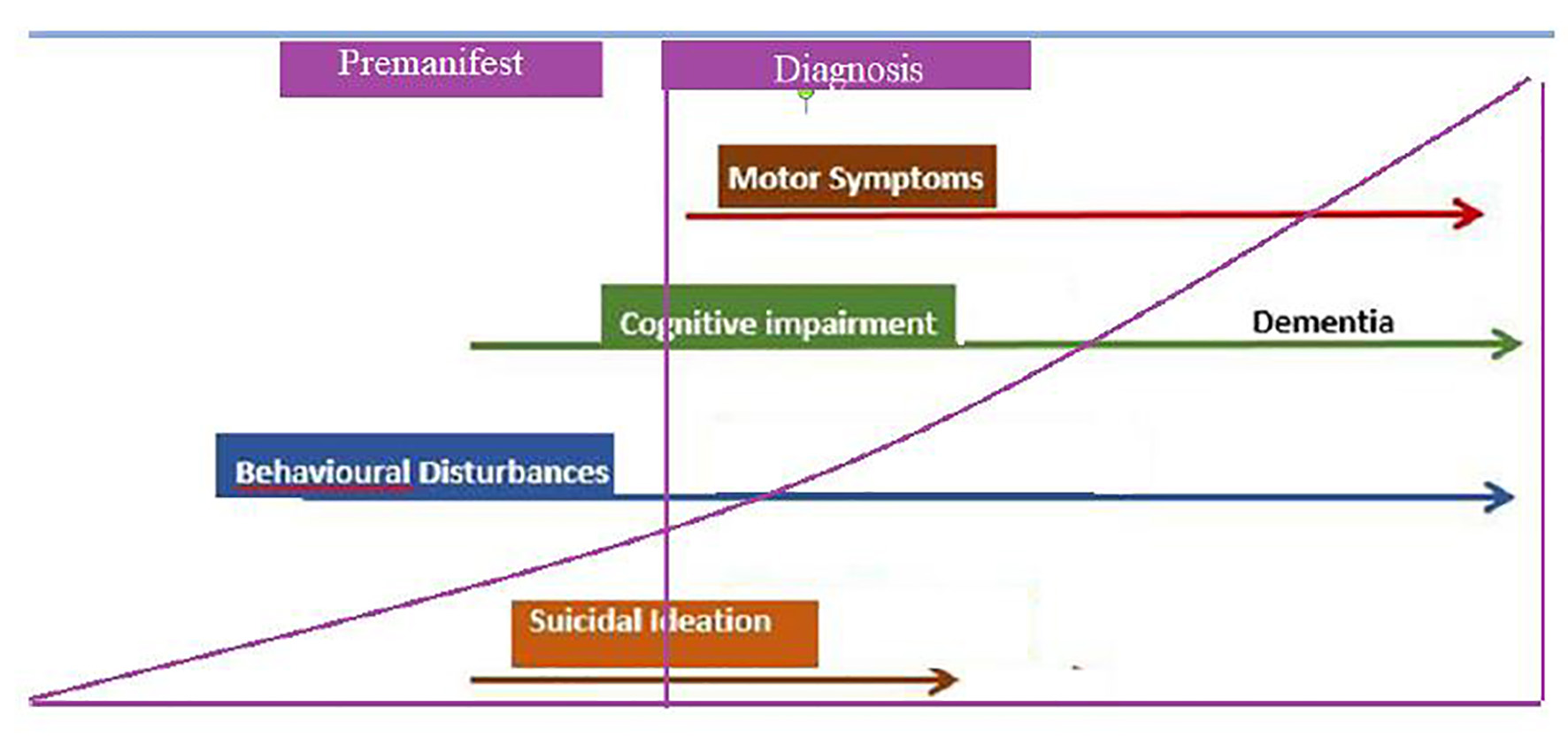 Click for large image | Figure 9. Natural evolution of Huntington’s disease. Functional capacity begins to decline, even from the premanifest stages, increasing motor disability. Regarding suicidal ideation, it is greater in premanifest stages, and begins to decrease as the disease progresses. |
Juvenile HD (JHD, Westphal variant)
JHD begins before the age of 20 years while childhood-onset begins before 10, both variants carry the largest CAG expansions (> 60 repeats). Its mean prevalence rate comes to 5%, probably underestimated due to the atypical presentation [34]. The clinical features are generally different from adults and much more varied, mostly characterized by parkinsonian signs and dystonia, aside from behavioral/mood and neuropsychiatric disturbances, learning disorders, as well as early and severe cognitive impairment (Westphal variant). In addition, childhood patients may also present cerebellar features, epilepsy, myoclonus, spasticity, developmental delay and autism, constituting a true challenge for an accurate diagnosis [35].
Fusilli et al reported the largest study in JHD. Two groups were identified, the long expansion (LE) group with a median CAG length of 61, and the highest expansion (HE) group with a median repeat size of 86. Although both shared common motor phenotypes, the HE subgroup showed a considerably lower age of onset (≤ 10 years of age) and increased mutation instability, manifesting with a wide variety of motor and non-motor symptoms along the disease course; they experienced a faster progression rate, reduced survival, and evidenced specific brain abnormalities not reported in patients with adult-onset HD [34]. The neuropsychiatric burden of JHD is considerably higher than adult forms, being the most impairing symptom when compared to motor features. Due to the low prevalence of the disease in this population and the great symptomatic heterogeneity, it is important for both clinicians and neurologists to take this into account based on the clinical history.
Prodromal HD
Subtle clinical features can manifest 10 - 15 years before motor symptoms. Since the development of the PREDICT-HD study, it was possible to establish the stage of prodromal HD and the estimated years to motor diagnosis using two variables: the number of CAG and the current age [36]. The prodromal period starts when any sign or symptom of HD is noted in a person whether at risk of developing HD or in the carrier group.
It is separated using a three-phase scheme [37]. In phase I or “low probability of diagnosis within 5 years” (< 60%), neuroimaging findings reveal brain volume loss, with the most prominent changes in the basal ganglia. Cognitive difficulties could be slightly present but not noticeable, with fatigue as a very common symptom that can be mistaken with apathy or depression.
In phase II or “medium probability of diagnosis within 5 years” (60-85%), approximate 7 - 13 years towards motor diagnosis, cognitive and behavioral problems might occur, as well as hyposmia [38]. Apathy, social cognition, verbal learning/memory, attention-information integration and sensory-perceptual processing are some domains of impairment in premanifest HD.
Finally, phase III or “high probability of diagnosis within 5 years” (> 85%) and “near motor diagnosis” (< 7 years), is the subgroup with the most marked rate of decline in all areas studied. Cognitive impairment becomes more evident with time, with complete lack of insight due to striato-frontal disruption and is much more disabling than the motor symptoms, involving a large family burden [39]. Within the cognitive sphere, the main areas affected are executive function, processing speed, working memory, and visuospatial control [40, 41].
Huntington’s disease integrated staging system (HD-ISS) is a valid system of classification which uses available clinical data to classify individuals into four stages. It comprises the whole HD course from birth (defined by the presence of the genetic expansion) to death. HD-ISS stage 0 includes pre-manifest individuals with ≥ 40 CAG repeats in the HTT gene. Stage 1 is verified with measurable indicators of underlying pathophysiology (biomarkers) but still asymptomatic. Stage 2 adds any sign or symptom to stage 1 and stage 3 includes symptomatic patients with loss of functionality in their activities of daily living [42].
Manifest HD
Motor features
Movement disorders are the main motor feature in HD. The disease is usually characterized by hyperkinetic movements that become hypokinetic in a later phase [33]. There are several scales to address motor function in HD, but the Unified Huntington Disease Rating Scale (UHDRS) is the most commonly used. The motor subset item in the UDHRS can be useful to assess clinical onset of HD.
Chorea is defined as an involuntary, spontaneous brief, rapid, purposeless, non-stereotyped, not rhythmic movement flowing randomly from one part of the body to another. In the initial stages of the disease, chorea affects appendicular and facial muscles (frontalis sign). As the disease progresses, it involves proximal muscles, affecting the stance and gait, leading to a bed-bidden condition. Swallowing becomes impaired with frequent choking events. During later stages, severe dysarthria and dysphagia are commonly seen, and patients could even become unable to speak. Additional findings include parakinesis (incorporation of the involuntary movement into a voluntary movement), motor impersistence (“milkmaid’s grip”) and hung-up reflexes.
Dystonia is a hyperkinetic movement characterized by sustained or intermittent muscle contractions that lead to repetitive movements, postures or both. It is usually worsened by action with overflow phenomenon. Dystonic features could be the initial motor manifestation in HD patients, such as cervical dystonia [43]. Other involuntary movements might be present including myoclonus and tics. Hypokinesia and/or akinesia are common components of the later stages in adult patients.
Cognitive features
Mild cognitive impairment (MCI) is present in more than 50% of premanifest HD patients. In cognitive assessment tests, Trail Making and Symbol digit show to be significantly altered in HD premanifest patients compared to controls. HD cognitive impairment has an initial impact on executive functions, mostly expressed as the difficulty with organization and mind flexibility. Language is relatively spared all throughout the disease. As disease progresses, subcortical and frontal dementia leads to fully dependency in all daily life activities [44].
In advanced stages, the pattern of dementia has been fully suggested as a differential diagnosis among HD, Alzheimer’s disease, and Parkinson’s disease dementia.
In 2010, HD Clinical Research Group at the University of California proposed a set of criteria to diagnose dementia in HD. Their results suggest that a diagnosis of HD dementia should include demonstrable evidence of impairment in at least two areas of cognition (e.g., attention, speed of processing, executive functions, visuospatial abilities, and memory), without necessarily having memory impairment expressed [45].
Behavior and psychiatric features
As for cognitive impairment, behavioral signs and symptoms usually precede motor features. It has been estimated that psychiatric manifestations are present in 33-76% of HD patients. Depression is the most frequent symptom, not quite related to disease stage. Suicide is more common in this group when compared to the general population, and more than 23% of HD patients have been described to have had at least one suicidal attempt. Suicide accounts for 3-7% of all HD deaths [46]. Other signs and symptoms often described are: anxiety (34-61%), irritability, apathy (70%), obsessions and hypersexuality. Psychosis is typically seen in later stages of the disease [1].
Autonomic disorders
People with HD have a higher incidence of falls. This is thought to be multifactorial, and although movement disorders such as chorea and parkinsonism aggravate this risk, autonomic dysfunction has been identified as one of the main contributors to falls in these patients.
Terroba-Chambi et al described a relationship between HD faller status and heart rate variability (HRV) in different postural positions, correlating the fall phenomenology with the activity of the autonomic nervous system. On the other hand, early sympathetic hyperactivity in the HD population appears associated to an increased rate of apoptosis-induced structural defect in the central autonomic network, such as the hypothalamus, the limbic system or the brainstem [47].
Hypertension is a modifiable cardiovascular risk factor implicated in several neurodegenerative disorders. There is a direct relationship between hypertension and HD severity, progression and clinical onset, while the use of antihypertensive drugs showed the opposite. These findings encourage further studies of the symptomatic or disease-modifying properties of antihypertensives in neurodegenerative diseases [48].
A recent review by Park et al showed how the circadian timing system (altered sleep/wake) may contribute to autonomic dysfunction in HD, with anterior hypothalamic involvement. Several preclinical models offered convincing evidence that the circadian timing system is compromised early in the disease process [49].
Moreover, the baroreceptor reflex dysfunction has been associated with orthostatism and risk of falling. This risk could be increased by the reduced HRV and supported by the intracellular inclusions in brainstem areas involved in autonomic regulation [50].
Oculomotor features
In symptomatic patients, oculomotor impairment includes apraxia of saccades, slow saccades, impersistence of gaze and distractibility. In premanifest HD, results remain controversial and variable according to the methodology employed (eye-tracking equipment or oculomotor items of the UHDRS). Some authors report horizontal ocular pursuit as the only oculomotor item of the UHDRS affected in premanifest HD [51].
| Differential Diagnosis | ▴Top |
HD accounts for 90-99% of patients who present the typical clinical picture of motor findings, cognitive decline and psychiatric disturbances, as mentioned above [52].
“HD phenocopy” is a term to describe those individuals who keep the clinical features of HD, without huntingtin mutation and irrespectively of the inherited pattern [53].
The new genetic technologies contribute to the growing number of HD-like disorders.
In Table 2, we present the most frequent phenocopies.
Click to view | Table 2. Differential Diagnoses of Huntington’s Disease, Autosomal Dominant (AD) and Autosomal Recessive (AR) Phenocopies, With Their Respective Genes Involved and Clinical Findings that Contribute to Reaching the Correct Diagnosis |
| Biomarkers | ▴Top |
Clinical biomarkers
Clinical biomarkers are standardized and protocolized clinical tests and rating scales that serve to assess the progression of motor, cognitive and psychiatric aspects of HD. These tests can be administered by personnel with requisite training and without need for expensive infrastructure, becoming accessible tool in most centers. However, they are not as sensitive as imaging biomarkers at 12-month follow-up. In this sense, PREDICT-HD and TRACK-HD studies provide great knowledge about clinical biomarkers including a Q- Motor assessment that represents an objective tool to detect early motor impairment, including tongue protrusion, forced grip and based tapping [54].
Neuroimaging biomarkers
Neuroimaging techniques include structural imaging and functional and metabolic imaging measures.
Structural imaging measures caudate nucleus and putamen atrophy as the most important and valuable aspect. These changes are best related with age of disease onset, CAG repeat length, and motor impairment. Additionally, white matter atrophy is another considerable finding of prodromal individuals as well as manifest patients, correlating proportionally with cognitive and motor decline. Cortical thinning has also been reported, and results suggest that individual variability in cortical volume loss at selected topographical regions might have a role in explaining the phenotypic variability. Finally, despite being less frequent and requiring special magnetic resonance sequences, metal deposition at the basal ganglia such as iron is another finding of the disease [55] (Fig. 10a, b).
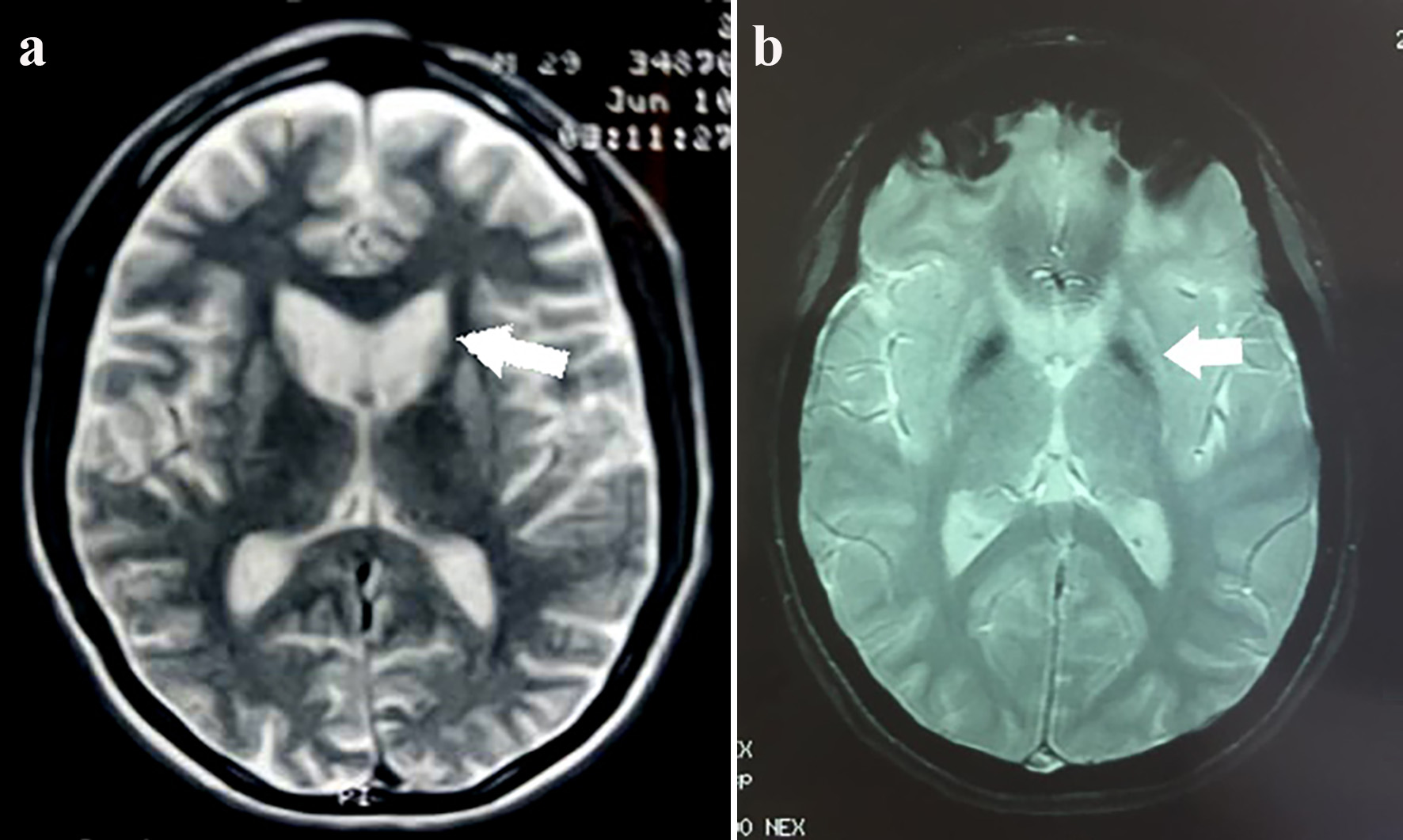 Click for large image | Figure 10. (a) Enlargement of both frontal horns of the lateral ventricles due to caudate atrophy (arrow). Axial T2-weighted sequence. (b) Diffuse bilateral and symmetric hypointense signals in T2 SWAN (magnetic susceptibility) sequence due to abnormal mineral deposition, slight dilation of the anterior extensions and lateral ventricles. Decreased volume of both globus pallidus (atrophy). |
Potential biomarkers could include measuring the total brain volume, as well as the striatum, caudate and putamen, cerebral white matter and ventricles. One of the limitations is that these biomarkers are susceptible to confounding factors that limit inter-study comparisons, such as technical differences. Finally, some inconstant results regarding the specificity and sensitivity of the measurements and their correlation with histological changes cast doubt on the usefulness of this method, and further studies are needed for it to be reliably used [56].
Macroscopic neurodegeneration in HD is likely to be preceded by substantial neuronal dysfunction many years beforehand. Hence, imaging techniques such as functional magnetic resonance imaging (fMRI), positron emission tomography (PET), and magnetic resonance spectroscopy (MRS) might allow identifying early neuronal physiological disturbances before noted in structural neuroimages. These bio-imaging techniques are better at monitoring early stages of disease progression and could even be used to measure treatment response [57] (Table 3).
Click to view | Table 3. Summary of Neuroimaging Biomarkers and Its Findings |
fMRI findings suggest that premanifest disease could be divided into at least two stages, regarding metabolic activity. An initial one, with predominant upregulation and increased regional brain activity, was followed by a later phase in which this level of activity decreases coinciding with the onset of clinical manifestations. In any case, these stages are still under investigation and need to be clarified through further longitudinal studies. The true functional and physiological importance of the altered fMRI activation patterns in this disorder is erratic and thus its use as a biomarker may prove to be difficult [58].
PET measures glucose reuptake and dopaminergic signalling. HD patients have been found to have a lower glucose metabolism at a striatal level and regional reductions in cortical glucose. The deficit at the caudate and cortical level is correlated with cognitive impairment, whereas striatal hypometabolism is more linked to motor deficits and reduced functional capacity [59].
Results from comparing PET imaging in manifest patients with presymptomatic gene carriers revealed a more marked deterioration rate of striatal metabolism in the former. Research studies of dopaminergic network, more specifically D1 and D2 receptors, have shown reduced concentration and activity of both in the striatum, in a directly proportional manner to the severity of the clinical findings and disease duration [60].
A longitudinal study with 18F-fluorodeoxyglucose PET (18FDG-PET) imaging in a premanifest group revealed a hypometabolism in certain regions, including the caudate and putamen, countered by relative hypermetabolism in thalamic structures at preclinical stages. These opposite mechanisms might indicate compensatory thalamic metabolic activity during a period of early neuronal loss, and the contrary was associated with the emergence of symptom onset. Despite everything stated above, the use of functional imaging as a biomarker will require further longitudinal studies comparing both groups, and is not yet available in clinical practice [61].
One of the most recent advances in functional imaging involves the intracellular enzyme phosphodiesterase 10A (PDE10A). This enzyme has a role in the regulation of striatal signalling with a high expression in the median spiny neurons of the striatum. The PDE10A inhibition improves cortico-basal function in HD with visible changes using a PDE10A PET radio ligand, although the Pfizer Amaryllis trial showed no symptomatic improvement [62]. As this specific protein suffers various modifications along the neurodegenerative process, it turned out to be a very valuable biomarker, showing alterations through PET imaging, even 15 to 20 years prior to clinical manifestations [63].
MRS recently became a novelty with possible profitability in HD. Lower concentrations of putaminal N-acetyl aspartate (marker of neuronal viability) have been found in premanifest and early disease patients compared to controls, proving early striatal neuronal loss [64]. Concerning its sensitivity compared to the aforementioned imaging studies, disease progression and response to treatment might correlate better with metabolite changes than with regional volumetric loss, indicating that MRS may be a superior method for measuring therapeutic efficacy. Controversial data were reported regarding striatal glutamate and glutamine concentrations using MRS technique in early stages. Both found to lead towards a potential excitotoxic state in HD [65].
Curiously, a supposed anti-excitotoxic role of creatine has been proposed, since the increase in intracellular energy levels after its supplementation attempted the decrease in the levels of glutamate and glutamine [66].
Larger samples are needed in order to prove whether this method is useful to determine disease evolution and response to treatment.
Biochemical biomarkers
Homeostasis alterations
In HD, there is a fail in maintaining homoeostasis, associated with increased physical stress, anxiety and mood disorders, which can be correlated with alterations inherent to the hypothalamic-pituitary-adrenal axis and the hypothalamic-pituitary-gonadal axis. Increased cortisol, adrenocorticotropic hormone and testosterone concentrations in plasma, serum and urine samples have been reported [67]. Nonetheless, these results have not been replicated thus far.
Endocrine disturbances
Weight loss leading to end-stage cachexia is commonly observed in some individuals, originally attributed to the caloric expenditure of choreic movements, but later studies observed that weight loss is also evident in patients with an akineto-rigid syndrome or even in pre-manifest cases, leading to the investigation of mechanisms that control weight, such as leptin, growth hormone, ghrelin and vasopressin, among others. As happened with other potential biomarkers, the evidence found was discrepant [68].
On the other hand, neuronal inclusions of HTT have been reported in the hypothalamus of HD patients involving neuroendocrine disturbances. Ahmad Aziz’s group showed that increases in cortisol production were mainly confined to the morning and early afternoon period, pointing toward a disturbed central glucocorticoid feedback regulation in HD patients due to an HPA axis dysfunction, becoming an early feature of the disease. Despite this, more studies are needed to confirm this hypothesis, and from it develop targeted therapies [69].
Mitochondrial dysfunction
Mitochondrial dysfunction is strongly implicated in the pathogenesis of HD. Therefore, several antioxidant compounds have shown promise as biomarker candidates, for example, decreased concentrations of glutathione peroxidase and copper-zinc superoxide dismutase in erythrocytes from HD patients compared with controls [70]. Higher concentrations of serum uric acid are associated with slower progression of the disease and progressive decreases in concentrations of creatine kinase were identified from both premanifest as manifest individuals [71].
Immune alterations
Investigations of the immune system in HD led to identified elevated cytokine concentrations, including interleukins 4, 6, 8, 10, and 23, tumor necrosis factor alpha (TNF-α) in brain and plasma samples of patients compared with controls [72]. Interleukin 6 and interleukin 8 levels showed similar concentrations between plasma and cerebrospinal fluid (CSF), with increasing values according to disease severity.
Combining different cytokines was more profitable at distinguishing disease states.
This was especially true regarding interleukin 6 concentration, which was increased in premanifest individuals at an average of 16 years from predicted phenoconversion, being the earliest biochemical abnormality identified in HD mutation carriers until now.
Although such compounds offer potential as biomarkers, increased immunological activity might be increased due to any other reason, such as comorbid conditions, commonly seen in this cohort [73].
Cholesterol metabolism
Oxidation of cholesterol at brain basis to 24S-hydoxy cholesterol (24OHC) is crucial for central nervous system development and its correct function and could be interrupted in HD. Low concentrations of 24OHC on plasma have been reported in patients and premanifest individuals, correlating with caudate atrophy. Not only was its metabolite found to be reduced, but also reductions in plasma concentrations of total cholesterol were reported in HD patients as well as in premanifest carriers compared with controls. Unfortunately, total cholesterol levels in first-degree relatives with negative genotype were also significantly lower than controls, suggesting unrelated genetic or environmental impacts. Nevertheless, 24OHC enables differentiation between healthy controls and premanifest individuals, indicating its potential as a biomarker [74].
Glutamate and GABAergic systems
Both glutamate and GABAergic systems, along with aminoacids such as aspartate and the enzymatic activity of aspartate and glutamate aminopeptidase, homovanilic acid, dopamine and prolactin have shown different results when comparing the findings between plasma and CSF, as well as between manifest and premanifest patients, making them unreliable biomarkers now [75, 76].
Neurofilaments (Nfs)
Nfs are heteropolymers composed by heavy, medium and light (NfL) chains and α-internexin subunits, that provide structural support for the axon caliber and intracellular traffic. They are released into the extracellular space and the CSF and, at lower concentrations, into the blood, depending on the degree of the axonal damage. Fourth-generation (single-molecule array) immunoassays can reliably measure both CSF and blood levels of Nf and are capable of detecting subtle changes along disease course. Patients with HD have shown increased Nf CSF levels, strongly correlated with disease progression. Therefore, Nf levels in blood could be included as a possible parameter in future clinical trials for this disease [77].
HD is a heterogeneous disease, and this influences the possible usefulness of the different biomarkers mentioned above. It is fairly evident that they tend to change during the course of the disease, rendering it unlikely to identify a single biomarker but rather a combination of them [78].
| Treatments | ▴Top |
Current available treatments are purely symptomatic and focus on improving motor and non-motor symptoms.
Many clinical trials have been working on the search for symptomatic and disease-modifying therapies. Furthermore, phase III human trials of gene silencing techniques are now ongoing.
The latest consensus about treatment options was performed by the European Huntington’s Disease Network (EHDN) and published in 2019. Data were obtained from 15 European experts from the national and steering committees and 73 worldwide additional experts from 25 countries. Here we will provide a summary of some of the aspects they mention [79] (Table 4).
Click to view | Table 4. Summary of the Most Commonly Used Drugs With Their Respective Doses in Each of the Symptoms Associated With Huntington’s Disease |
Symptomatic Therapy
Motor symptoms
Chorea only requires treatment if it impacts or impairs the patient’s quality of life, functionality, or safety. The efficacy of typical neuroleptics is well known; however, these drugs have many adverse effects such as tardive dyskinesia, akathisia, acute dystonia, parkinsonism, sedation and depression. Atypical neuroleptics (clozapine, quetiapine, risperidone, olanzapine, and aripiprazole), on the other hand, are potentially effective with less incidence of tardive extrapyramidal syndromes, mainly regarding clozapine and quetiapine, but recently cognitive impairment has been reported associated with their chronic use [79].
In juvenile forms, dopamine agonists such as pramipexole (PMX) showed improvement in motor and depression scales compared to levodopa, but considering their behavioral adverse effects, they are not widely recommended. A recent work of Ravello et al showed a possible neuroprotective role of PMX in HD. These statements are based on the fact that autophagy can be promoted from the stimulation of D2-D3 receptors, encouraging htt clearance. They tested this drug on mice for 4 weeks and registered reduced striatal levels of soluble HTT afterwards [80]. It is not discarded that improvement produced by PMX could be due to direct dopaminergic/symptomatic effects on motor regulation rather than to neuroprotection and further investigation is needed.
Tetrabenazine (TBZ) is the only medication approved for treatment of chorea in HD. It selectively depletes central monoamines by reversibly binding to the type 2 vesicular monoamine transporter (VMAT-2). Since VMAT-2 contains and transports serotonin, dopamine, and norepinephrine from the cytoplasm into presynaptic vesicles, its inhibition leads to early degradation of these monoamines. The reduction of dopamine improves chorea, while depletion of serotonin and norepinephrine may result in worsening depression and anxiety, and increase the risk of suicide [81].
Two new drugs were recently developed and approved for the treatment of chorea. Deutetrabenazine is a deuterated TBZ with a longer half-life (two daily dosing) and better tolerability than TBZ. Valbenazine, a prodrug of an isomer of TBZ, with a longer half-life allowing once-a-day dosing, previously approved for tardive dyskinesia, is under FDA submission for chorea [82].
Regarding motor symptoms other than chorea, there are many symptomatic options that vary according to each one. Unluckily, these options have limited benefits and are based on low-quality evidence. Some examples are the use of levodopa and dopamine agonists in Westphal variant for parkinsonian and dystonic features, valproic acid or benzodiazepines for myoclonus [83] and botulinum toxin in patients with severe or disabling focal or segmental dystonia.
Cognitive symptoms
Based on a significant loss of acetylcholine and acetylcholine transferase activity in the striatum, nucleus accumbens and hippocampus in HD patients, rivastigmine has been evaluated in some studies. The results are variable but currently it is still the best available option at treating cognitive symptoms.
One of the theories postulated asserts that at least part of the neurodegeneration in HD occurs as a result of glutamatergic excitotoxicity in the striatum. One small, open-label study suggested a potential neuroprotective effect following long-term treatment with memantine, a non-competitive glutamate receptor antagonist; however, this finding still needs confirmation and memantine is currently not approved for treatment of cognitive impairment in HD [84].
Psychiatric symptoms
Depression is the most common and disabling psychiatric symptom (33-70%). There is a known benefit of the selective serotonin reuptake inhibitors (SSRIs), especially fluoxetine and citalopram. In cases of SSRI intolerance, tricyclic antidepressants such as nortriptyline or desipramine may be used, whose resulting anticholinergic capacity is less than amitriptyline [85]. In cases of refractory depression, atypical antipsychotics such as olanzapine, risperidone, aripiprazole or clozapine have all shown benefit in case reports and small open-label case series [86-88].
SSRIs are also useful to treat obsessive and/or compulsive symptoms, very commonly seen among this population. In the case that adjunctive therapy is needed, the recommended drugs are those mood-stabilizing antiepileptic drugs (AEDs), including valproate, carbamazepine, lamotrigine and topiramate, all with variable benefits among patients [89].
Agitation may respond to benzodiazepines such as clonazepam. Pharmacological treatment of irritability includes SSIRs (fluoxetine, sertraline) as first-line therapy, and clomipramine and buspirone as second-choice strategies [79].
Curiously, only a few clinical trials have been conducted in behavioral disorders, and recent research failed to demonstrate a benefit of buspirone in treatment of apathy. Ongoing studies are aiming to evaluate dextromethorphan/quinidine and a vasopressin 1A receptor antagonist (SRX46) for irritability.
Psychosis and delusions are less common symptoms and have been reported in 3-11% of patients, with more extensiveness in later stages of the disease. In these cases, atypical antipsychotics are recommended [90].
Cannabis in HD
In recent decades, the endocannabinoid system has attracted considerable interest as a possible therapeutic target in numerous pathological conditions. Its involvement in various physiological processes is well known, such as energy balance, appetite stimulation, blood pressure, pain modulation, nausea and vomiting control, memory, learning and immunity, as well as in pathological conditions where it plays a protective role. It has been reported that changes in endocannabinoid levels may be related to some neurodegenerative diseases such as Parkinson’s disease, Alzheimer’s disease, and H’D.
Numerous preclinical studies have described altered endocannabinoid system (ECS) expression in the areas involved in HD, suggesting a role in disease progression. Post-mortem studies of HD patients showed a significant loss of CB1 receptors (nearly 97.5%) in basal ganglia structures, especially in the globus pallidus. Despite this, the administration of oral cannabidiol (CBD) (10 mg/kg/day for 6 weeks) did not improve any of the symptoms of patients with HD, and the use of nabilone enhanced chorea and cognitive problems [91]. More studies are needed to establish the potential use of cannabinoids in HD.
Role of neuroglobin (Ngb)
Ngb is a 17-kDa monomeric hexa-coordinated heme protein belonging to the globin family, of which until now only hemoglobin (Hb) and myoglobin (Mb) were known to be present in vertebrates [92]. Ngb was discovered in the year 2000, and was found to be highly conserved throughout evolution. It is extensively expressed in both central and peripheral nervous systems. Higher levels of Ngb were found in hypoxic-ischemic pathologies, from which its possible neuroprotective role was inferred. It has been reported that Ngb overexpression is related to cytoprotective effects on neurons, anti-apoptotic features on nerve tissue and protection against oxidative stress [93].
The main physiological functions of Ngb are binding and transport of O2, removing reactive species. The potential neuroprotective role was investigated in many neurodegenerative diseases such as Alzheimer’s disease and HD [15]. Apparently, HTT and Ngb have a neuroprotective role triggered by the hormone 17β-estradiol (E2) protecting neurons from apoptosis, and this synergic pathway seems to fail when huntingtin is mutated. The Ngb neuroprotective role is probably related to a synergic mechanism which involves improving mitochondria function, decreasing the secretion of reactive oxygen species and nitric oxygen and inhibiting the intrinsic pathway of cell death.
Considering the aforementioned mechanisms, inducing the Ngb expression in different pathologies, such as ischemia, hypoxia, AD and HD, could be a new therapeutic approach against neurodegenerative diseases [94].
Unluckily, recombinant Ngb therapy is not suitable as it is an intracellular protein which does not cross cellular membranes. However, small molecules (natural and synthetic) proved well as vectors which overpass blood brain barrier, inducing the upregulation of Ngb both in vitro and in vivo experiments [95]. Several molecules able to induce Ngb are under study, such as iron chelators (deferoxamine), hormones (estrogens), short-chain fatty acid derivatives (cinnamic and valproic acids) nonsteroidal anti-inflammatory drugs and metformin [96].
So much material is available regarding Ngb and its neuroprotective role, and the induced overexpression could represent a possible therapeutic approach to treat neurodegenerative diseases that, to date, are lacking in effective therapies.
Disease-modifying approaches: targeting mutant huntingtin
Antisense oligonucleotides (ASOs)
ASOs are capable to reduce, restore, or modify RNA and protein expression. ASOs are synthetic single-stranded DNA analogs, usually 16 to 22 bases long, that selectively bind to specific complementary disease-causing pre-messenger RNA (mRNA), previous chemical modifications to avoid rapid degradation by cellular nucleases.
ASOs can regulate target gene expression in different ways, most commonly by recruiting ribonuclease H1 (RNase H1). After selective binding of the ASO to its target RNA, an RNA-DNA hybrid is formed, inducing messenger RNA (mRNA) degradation by RNase H1.
The ASO-based therapeutic trial using an HTT-targeting non-selective ASO RG6042 has been halted for futility. A second one, called Precision-HD1 uses the allele-specific ASOs WVE-120101 and WVE-120102, targeting mutant htt mRNA, preserving the translation of the healthy allele [97]. Low levels of HTT were maintained even after a single intrathecal injection of ASO in animal models, suggesting a prolonged but reversible effect in HD patients [98]. Preliminary reports showed intrathecal delivery of RG6042 ASO to be safe and well tolerated, although cases of thrombocytopenia were reported in some patients, making it a parameter to monitor during the study. ASOs that target specific single-nucleotide polymorphisms linked to the CAG expansion show promise in preclinical models of HD. Unfortunately, many of these ASO therapies failed to reach the primary end point and new post-hoc analyses are under review.
Targeting DNA
Currently, approaches targeting DNA are aimed at modulating the CAG repeats. They can either modulate gene transcription or directly alter the HTT gene by using specific DNA-binding elements. Ongoing trials are focused on three main classes of nucleases designed for DNA-targeting purposes: zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and Cas9 or other RNA-guided bacterial nucleases, each of them with a different mode of action and DNA binding mechanism.
1) ZFNs
ZFNs are active effectors nucleases, linked to a DNA binding element built of an assemblage of multiple zinc finger peptides. Each one of them can bind a sequence of three to five different nucleotides of the DNA chain. Zinc-finger proteins (ZFPs) without nuclease activity can lower gene expression just by binding to the DNA strand and precluding gene transcription. Regarding its possible use in HD, ZFPs can be designed to bind selectively to expanded CAG repeats without altering normal ones. Recent data showed lower HTT expression by ZFPs, with no impact on normal proteins. These results encourage further studies in order to develop allele-specific ZFP repressors and ZFNs as potential therapies for HD [99].
2) TALENs
TALENs are very similar to ZFNs. They use a nuclease effector domain bound to a DNA recognition domain. This binomial uses a series of specific repetitions of amino acids that bind to a specific nucleotide, with different amino acid combinations recognizing different DNA sequences. They have shown higher efficiency and specificity compared to ZFN, but as they need a specific nucleotide at the end of the DNA sequence, it could be a limitation with potential targets. The only work published on HD patients using TALENs derived fibroblasts showed very low efficiency, encouraging further studies in this area [99].
3) CRISPR/Cas9
CRISPR/Cas9 system is based on a bacterial immune system that recognizes and destroys foreign DNA. The acronym that gives its name stands for “clustered regularly interspaced short palindromic repeats”, and the Cas9 protein is an RNA-guided nuclease that cleaves double-strand breaks in specific DNA sites. Unlike ZFNs or TALENs, this system does not use a DNA recognition domain, but rather specific RNA machinery that targets the DNA areas of interest [99].
Small molecules
Small molecules targeting RNA is an orally bioavailable drug that crosses the blood brain barrier and modulates the splicing of HTT precursor mRNA. It is a promising disease-modifying strategy as they have shown to reduce HTT levels in HD patient-derived skin cells and neurons, leading to a 50% reduction of CSF HTT in two mouse models trials [99]. Human trials are ongoing.
Other additional targets to mention include sigma receptors agonist (pridopridine) involved in several mechanisms, such as neuroinflammation modulation through the complement cascade (specifically C1Q), gene therapy conversion of striatal astrocytes into GABAergic neurons. Probably, one of the most promising targets is a new ASO which targets MSH3, one of the components of the DNA repairs pathways acting as HTT modifier.
| Conclusions | ▴Top |
HD is a progressive and devastating disorder for both the individual and the family. It affects those who suffer from it not only physically, but also psychologically and socially. A vast number of publications have come to light improving our knowledge about the disease, from its natural history and pathogenesis as well as the care for patients, which fortunately has increased over the past two decades.
So where do we stand regarding HD and where are we headed? Many studies are based on further understanding its pathophysiology and finding a reliable biomarker that can be easily reproduced. This may be path to developing the long-awaited drug that interferes with the disease process. To date, few treatments are available and a large number of clinical trials have failed. However, the ongoing development of new therapeutic strategies capable of targeting HTT directly announces a new era for HD investigation and we believe we are closer than ever to finding true potential treatments to modify and even prevent HD. The developments are promising but there is still a long way to go.
Acknowledgments
We thank all the participants in this review for fostering enriching discussions.
Financial Disclosure
None to declare.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Author Contributions
Natalia Gonzalez Rojas: conception, organization, and execution of research project, and writing of the first draft of manuscript. Martin Emiliano Cesarini: conception and execution of research project. Guillermo Peker: conception and execution of research project. Gustavo Andres Da Prat: execution of research project. Jose Luis Etcheverry: organization of research project. Emilia Mabel Gatto: conception, organization, and execution of research project, and review and critique of manuscript.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Roos RA. Huntington's disease: a clinical review. Orphanet J Rare Dis. 2010;5:40.
doi pubmed - Rawlins MD, Wexler NS, Wexler AR, Tabrizi SJ, Douglas I, Evans SJ, Smeeth L. The Prevalence of Huntington's Disease. Neuroepidemiology. 2016;46(2):144-153.
doi pubmed - Castilhos RM, Augustin MC, Santos JA, Perandones C, Saraiva-Pereira ML, Jardim LB, Rede N. Genetic aspects of Huntington's disease in Latin America. A systematic review. Clin Genet. 2016;89(3):295-303.
doi pubmed - Fisher ER, Hayden MR. Multisource ascertainment of Huntington disease in Canada: prevalence and population at risk. Mov Disord. 2014;29(1):105-114.
doi pubmed - Xu M, Wu ZY. Huntington Disease in Asia. Chin Med J (Engl). 2015;128(13):1815-1819.
doi pubmed - A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell. 1993;72(6):971-983.
doi - Cattaneo E, Zuccato C, Tartari M. Normal huntingtin function: an alternative approach to Huntington's disease. Nat Rev Neurosci. 2005;6(12):919-930.
doi pubmed - Nance MA. Genetics of Huntington disease. Handb Clin Neurol. 2017;144:3-14.
doi pubmed - Bunting EL, Hamilton J, Tabrizi SJ. Polyglutamine diseases. Curr Opin Neurobiol. 2022;72:39-47.
doi pubmed - Nopoulos PC. Huntington disease: a single-gene degenerative disorder of the striatum. Dialogues Clin Neurosci. 2016;18(1):91-98.
doi pubmed - Rosenblatt A, Brinkman RR, Liang KY, Almqvist EW, Margolis RL, Huang CY, Sherr M, et al. Familial influence on age of onset among siblings with Huntington disease. Am J Med Genet. 2001;105(5):399-403.
doi pubmed - Chattopadhyay B, Baksi K, Mukhopadhyay S, Bhattacharyya NP. Modulation of age at onset of Huntington disease patients by variations in TP53 and human caspase activated DNase (hCAD) genes. Neurosci Lett. 2005;374(2):81-86.
doi pubmed - Wexler NS, Lorimer J, Porter J, Gomez F, Moskowitz C, Shackell E, Marder K, et al. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington's disease age of onset. Proc Natl Acad Sci U S A. 2004;101(10):3498-3503.
doi pubmed - Gusella JF, MacDonald ME. Huntington's disease: the case for genetic modifiers. Genome Med. 2009;1(8):80.
doi pubmed - Gusella JF, MacDonald ME, Lee JM. Genetic modifiers of Huntington's disease. Mov Disord. 2014;29(11):1359-1365.
doi pubmed - Bakels HS, Roos RAC, van Roon-Mom WMC, de Bot ST. Juvenile-Onset Huntington Disease Pathophysiology and Neurodevelopment: A Review. Mov Disord. 2022;37(1):16-24.
doi pubmed - Uhlmann WR, Penaherrera MS, Robinson WP, Milunsky JM, Nicholson JM, Albin RL. Biallelic mutations in huntington disease: A new case with just one affected parent, review of the literature and terminology. Am J Med Genet A. 2015;167A(5):1152-1160.
doi pubmed - Ciosi M, Maxwell A, Cumming SA, Hensman Moss DJ, Alshammari AM, Flower MD, Durr A, et al. A genetic association study of glutamine-encoding DNA sequence structures, somatic CAG expansion, and DNA repair gene variants, with Huntington disease clinical outcomes. EBioMedicine. 2019;48:568-580.
doi pubmed - Gatto EM, Rojas NG, Persi G, Etcheverry JL, Cesarini ME, Perandones C. Huntington disease: Advances in the understanding of its mechanisms. Clin Park Relat Disord. 2020;3:100056.
doi pubmed - Sathasivam K, Neueder A, Gipson TA, Landles C, Benjamin AC, Bondulich MK, Smith DL, et al. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc Natl Acad Sci U S A. 2013;110(6):2366-2370.
doi pubmed - Wellington CL, Leavitt BR, Hayden MR. Huntington disease: new insights on the role of huntingtin cleavage. J Neural Transm Suppl. 2000;58:1-17.
doi pubmed - Hervas-Corpion I, Guiretti D, Alcaraz-Iborra M, Olivares R, Campos-Caro A, Barco A, Valor LM. Early alteration of epigenetic-related transcription in Huntington's disease mouse models. Sci Rep. 2018;8(1):9925.
doi pubmed - Francelle L, Lotz C, Outeiro T, Brouillet E, Merienne K. Contribution of Neuroepigenetics to Huntington's Disease. Front Hum Neurosci. 2017;11:17.
doi pubmed - Krol J, Fiszer A, Mykowska A, Sobczak K, de Mezer M, Krzyzosiak WJ. Ribonuclease dicer cleaves triplet repeat hairpins into shorter repeats that silence specific targets. Mol Cell. 2007;25(4):575-586.
doi pubmed - Gunawardena S, Her LS, Brusch RG, Laymon RA, Niesman IR, Gordesky-Gold B, Sintasath L, et al. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron. 2003;40(1):25-40.
doi - Wong YC, Holzbaur EL. The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J Neurosci. 2014;34(4):1293-1305.
doi pubmed - Harding RJ, Tong YF. Proteostasis in Huntington's disease: disease mechanisms and therapeutic opportunities. Acta Pharmacol Sin. 2018;39(5):754-769.
doi pubmed - Martin DD, Ladha S, Ehrnhoefer DE, Hayden MR. Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci. 2015;38(1):26-35.
doi pubmed - Carmo C, Naia L, Lopes C, Rego AC. Mitochondrial dysfunction in Huntington's Disease. Adv Exp Med Biol. 2018;1049:59-83.
doi pubmed - Guo JL, Lee VM. Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat Med. 2014;20(2):130-138.
doi pubmed - Bradford J, Shin JY, Roberts M, Wang CE, Sheng G, Li S, Li XJ. Mutant huntingtin in glial cells exacerbates neurological symptoms of Huntington disease mice. J Biol Chem. 2010;285(14):10653-10661.
doi pubmed - Di Pardo A, Pepe G, Castaldo S, Marracino F, Capocci L, Amico E, Madonna M, et al. Stimulation of Sphingosine Kinase 1 (SPHK1) is beneficial in a Huntington's disease pre-clinical model. Front Mol Neurosci. 2019;12:100.
doi pubmed - Ghosh R, Tabrizi SJ. Clinical Features of Huntington's Disease. Adv Exp Med Biol. 2018;1049:1-28.
doi pubmed - Fusilli C, Migliore S, Mazza T, Consoli F, De Luca A, Barbagallo G, Ciammola A, et al. Biological and clinical manifestations of juvenile Huntington's disease: a retrospective analysis. Lancet Neurol. 2018;17(11):986-993.
doi - Nopoulos PC. Special issue: juvenile onset Huntington's disease. Brain Sci. 2020;10(9):652.
doi pubmed - Paulsen JS, Smith MM, Long JD, investigators PH, Coordinators of the Huntington Study Group. Cognitive decline in prodromal Huntington Disease: implications for clinical trials. J Neurol Neurosurg Psychiatry. 2013;84(11):1233-1239.
doi pubmed - Tabrizi SJ, Scahill RI, Owen G, Durr A, Leavitt BR, Roos RA, Borowsky B, et al. Predictors of phenotypic progression and disease onset in premanifest and early-stage Huntington's disease in the TRACK-HD study: analysis of 36-month observational data. Lancet Neurol. 2013;12(7):637-649.
doi - Paulsen DJ, Hallquist MN, Geier CF, Luna B. Effects of incentives, age, and behavior on brain activation during inhibitory control: a longitudinal fMRI study. Dev Cogn Neurosci. 2015;11:105-115.
doi pubmed - Cardoso F. Nonmotor symptoms in Huntington disease. Int Rev Neurobiol. 2017;134:1397-1408.
doi pubmed - Harrington DL, Rubinov M, Durgerian S, Mourany L, Reece C, Koenig K, Bullmore E, et al. Network topology and functional connectivity disturbances precede the onset of Huntington's disease. Brain. 2015;138(Pt 8):2332-2346.
doi pubmed - Kerschbamer E, Biagioli M. Huntington's disease as neurodevelopmental disorder: altered chromatin regulation, coding, and non-coding RNA transcription. Front Neurosci. 2015;9:509.
doi pubmed - Tabrizi SJ, Schobel S, Gantman EC, Mansbach A, Borowsky B, Konstantinova P, Mestre TA, et al. A biological classification of Huntington's disease: the Integrated Staging System. Lancet Neurol. 2022;21(7):632-644.
doi - Jinnah HA, Albanese A. The new classification system for the dystonias: why was it needed and how was it developed? Mov Disord Clin Pract. 2014;1(4):280-284.
doi pubmed - Dumas EM, van den Bogaard SJ, Middelkoop HA, Roos RA. A review of cognition in Huntington's disease. Front Biosci (Schol Ed). 2013;5(1):1-18.
doi pubmed - Peavy GM, Jacobson MW, Goldstein JL, Hamilton JM, Kane A, Gamst AC, Lessig SL, et al. Cognitive and functional decline in Huntington's disease: dementia criteria revisited. Mov Disord. 2010;25(9):1163-1169.
doi pubmed - Huntington Study Group Cohort Investigators, Dorsey E. Characterization of a large group of individuals with huntington disease and their relatives enrolled in the COHORT study. PLoS One. 2012;7(2):e29522.
doi pubmed - Terroba-Chambi C, Bruno V, Vigo DE, Merello M. Heart rate variability and falls in Huntington's disease. Clin Auton Res. 2021;31(2):281-292.
doi pubmed - Mancia G, Grassi G. The autonomic nervous system and hypertension. Circ Res. 2014;114(11):1804-1814.
doi pubmed - Park S, Colwell CS. Do disruptions in the circadian timing system contribute to autonomic dysfunction in Huntington's disease? Yale J Biol Med. 2019;92(2):291-303.
- Bar KJ, Boettger MK, Andrich J, Epplen JT, Fischer F, Cordes J, Koschke M, et al. Cardiovagal modulation upon postural change is altered in Huntington's disease. Eur J Neurol. 2008;15(8):869-871.
doi pubmed - Winder JY, Roos RAC. Premanifest Huntington's disease: Examination of oculomotor abnormalities in clinical practice. PLoS One. 2018;13(3):e0193866.
doi pubmed - Cardoso F. Differential diagnosis of Huntington's disease: what the clinician should know. Neurodegener Dis Manag. 2014;4(1):67-72.
doi pubmed - Hensman Moss DJ, Poulter M, Beck J, Hehir J, Polke JM, Campbell T, Adamson G, et al. C9orf72 expansions are the most common genetic cause of Huntington disease phenocopies. Neurology. 2014;82(4):292-299.
doi pubmed - Tabrizi SJ, Scahill RI, Durr A, Roos RA, Leavitt BR, Jones R, Landwehrmeyer GB, et al. Biological and clinical changes in premanifest and early stage Huntington's disease in the TRACK-HD study: the 12-month longitudinal analysis. Lancet Neurol. 2011;10(1):31-42.
doi - Aylward EH, Li Q, Stine OC, Ranen N, Sherr M, Barta PE, Bylsma FW, et al. Longitudinal change in basal ganglia volume in patients with Huntington's disease. Neurology. 1997;48(2):394-399.
doi pubmed - Aylward EH, Sparks BF, Field KM, Yallapragada V, Shpritz BD, Rosenblatt A, Brandt J, et al. Onset and rate of striatal atrophy in preclinical Huntington disease. Neurology. 2004;63(1):66-72.
doi pubmed - Kloppel S, Henley SM, Hobbs NZ, Wolf RC, Kassubek J, Tabrizi SJ, Frackowiak RS. Magnetic resonance imaging of Huntington's disease: preparing for clinical trials. Neuroscience. 2009;164(1):205-219.
doi pubmed - Saft C, Schuttke A, Beste C, Andrich J, Heindel W, Pfleiderer B. fMRI reveals altered auditory processing in manifest and premanifest Huntington's disease. Neuropsychologia. 2008;46(5):1279-1289.
doi pubmed - Backman L, Robins-Wahlin TB, Lundin A, Ginovart N, Farde L. Cognitive deficits in Huntington's disease are predicted by dopaminergic PET markers and brain volumes. Brain. 1997;120(Pt 12):2207-2217.
doi pubmed - Pavese N, Andrews TC, Brooks DJ, Ho AK, Rosser AE, Barker RA, Robbins TW, et al. Progressive striatal and cortical dopamine receptor dysfunction in Huntington's disease: a PET study. Brain. 2003;126(Pt 5):1127-1135.
doi pubmed - Fazio P, Paucar M, Svenningsson P, Varrone A. Novel imaging biomarkers for Huntington's disease and other hereditary choreas. Curr Neurol Neurosci Rep. 2018;18(12):85.
doi pubmed - https://clinicaltrials.gov/ct2/show/NCT02197130.
- Niccolini F, Haider S, Reis Marques T, Muhlert N, Tziortzi AC, Searle GE, Natesan S, et al. Altered PDE10A expression detectable early before symptomatic onset in Huntington's disease. Brain. 2015;138(Pt 10):3016-3029.
doi pubmed - Sturrock A, Laule C, Decolongon J, Dar Santos R, Coleman AJ, Creighton S, Bechtel N, et al. Magnetic resonance spectroscopy biomarkers in premanifest and early Huntington disease. Neurology. 2010;75(19):1702-1710.
doi pubmed - Sanchez-Pernaute R, Garcia-Segura JM, del Barrio Alba A, Viano J, de Yebenes JG. Clinical correlation of striatal 1H MRS changes in Huntington's disease. Neurology. 1999;53(4):806-812.
doi pubmed - Bender A, Auer DP, Merl T, Reilmann R, Saemann P, Yassouridis A, Bender J, et al. Creatine supplementation lowers brain glutamate levels in Huntington's disease. J Neurol. 2005;252(1):36-41.
doi pubmed - Saleh N, Moutereau S, Durr A, Krystkowiak P, Azulay JP, Tranchant C, Broussolle E, et al. Neuroendocrine disturbances in Huntington's disease. PLoS One. 2009;4(3):e4962.
doi pubmed - Popovic V, Svetel M, Djurovic M, Petrovic S, Doknic M, Pekic S, Miljic D, et al. Circulating and cerebrospinal fluid ghrelin and leptin: potential role in altered body weight in Huntington's disease. Eur J Endocrinol. 2004;151(4):451-455.
doi pubmed - Aziz NA, Pijl H, Frolich M, van der Graaf AW, Roelfsema F, Roos RA. Increased hypothalamic-pituitary-adrenal axis activity in Huntington's disease. J Clin Endocrinol Metab. 2009;94(4):1223-1228.
doi pubmed - Mochel F, Charles P, Seguin F, Barritault J, Coussieu C, Perin L, Le Bouc Y, et al. Early energy deficit in Huntington disease: identification of a plasma biomarker traceable during disease progression. PLoS One. 2007;2(7):e647.
doi pubmed - Kim J, Amante DJ, Moody JP, Edgerly CK, Bordiuk OL, Smith K, Matson SA, et al. Reduced creatine kinase as a central and peripheral biomarker in Huntington's disease. Biochim Biophys Acta. 2010;1802(7-8):673-681.
doi pubmed - Bjorkqvist M, Wild EJ, Thiele J, Silvestroni A, Andre R, Lahiri N, Raibon E, et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington's disease. J Exp Med. 2008;205(8):1869-1877.
doi pubmed - Wild E, Bjorkqvist M, Tabrizi SJ. Immune markers for Huntington's disease? Expert Rev Neurother. 2008;8(12):1779-1781.
doi pubmed - Markianos M, Panas M, Kalfakis N, Vassilopoulos D. Low plasma total cholesterol in patients with Huntington's disease and first-degree relatives. Mol Genet Metab. 2008;93(3):341-346.
doi pubmed - Stahl SM, Thiemann S, Faull KF, Barchas JD, Berger PA. Neurochemistry of dopamine in Huntington's dementia and normal aging. Arch Gen Psychiatry. 1986;43(2):161-164.
doi pubmed - Markianos M, Panas M, Kalfakis N, Vassilopoulos D. Plasma homovanillic acid and prolactin in Huntington's disease. Neurochem Res. 2009;34(5):917-922.
doi pubmed - Khalil M, Teunissen CE, Otto M, Piehl F, Sormani MP, Gattringer T, Barro C, et al. Neurofilaments as biomarkers in neurological disorders. Nat Rev Neurol. 2018;14(10):577-589.
doi pubmed - Kloppel S, Chu C, Tan GC, Draganski B, Johnson H, Paulsen JS, Kienzle W, et al. Automatic detection of preclinical neurodegeneration: presymptomatic Huntington disease. Neurology. 2009;72(5):426-431.
doi pubmed - Bachoud-Levi AC, Ferreira J, Massart R, Youssov K, Rosser A, Busse M, Craufurd D, et al. International guidelines for the treatment of Huntington's disease. Front Neurol. 2019;10:710.
doi pubmed - Luis-Ravelo D, Estevez-Silva H, Barroso-Chinea P, Afonso-Oramas D, Salas-Hernandez J, Rodriguez-Nunez J, Acevedo-Arozena A, et al. Pramipexole reduces soluble mutant huntingtin and protects striatal neurons through dopamine D3 receptors in a genetic model of Huntington's disease. Exp Neurol. 2018;299(Pt A):137-147.
doi pubmed - Hayden MR, Leavitt BR, Yasothan U, Kirkpatrick P. Tetrabenazine. Nat Rev Drug Discov. 2009;8(1):17-18.
doi pubmed - Bashir H, Jankovic J. Treatment options for chorea. Expert Rev Neurother. 2018;18(1):51-63.
doi pubmed - Saft C, Lauter T, Kraus PH, Przuntek H, Andrich JE. Dose-dependent improvement of myoclonic hyperkinesia due to Valproic acid in eight Huntington's Disease patients: a case series. BMC Neurol. 2006;6:11.
doi pubmed - Li Y, Hai S, Zhou Y, Dong BR. Cholinesterase inhibitors for rarer dementias associated with neurological conditions. Cochrane Database Syst Rev. 2015;3:CD009444.
doi - Mendez MF. Mania in neurologic disorders. Curr Psychiatry Rep. 2000;2(5):440-445.
doi pubmed - Carr LA, Perlman S. Risperidone and olanzapine improve motor symptoms in Huntington's disease. Neurol Suppl. 2000;3:A116.
- Ciammola A, Sassone J, Colciago C, Mencacci NE, Poletti B, Ciarmiello A, Squitieri F, et al. Aripiprazole in the treatment of Huntington's disease: a case series. Neuropsychiatr Dis Treat. 2009;5:1-4.
doi - Sajatovic M, Verbanac P, Ramirez LF, Meltzer HY. Clozapine treatment of psychiatric symptoms resistant to neuroleptic treatment in patients with Huntington's chorea. Neurology. 1991;41(1):156.
doi pubmed - Patzold T, Brune M. Obsessive compulsive disorder in huntington disease: a case of isolated obsessions successfully treated with sertraline. Neuropsychiatry Neuropsychol Behav Neurol. 2002;15(3):216-219.
- Alpay M, Koroshetz WJ. Quetiapine in the treatment of behavioral disturbances in patients with Huntington's disease. Psychosomatics. 2006;47(1):70-72.
doi pubmed - Fraguas-Sanchez AI, Torres-Suarez AI. Medical use of cannabinoids. Drugs. 2018;78(16):1665-1703.
doi pubmed - Pesce A, Bolognesi M, Bocedi A, Ascenzi P, Dewilde S, Moens L, Hankeln T, et al. Neuroglobin and cytoglobin. Fresh blood for the vertebrate globin family. EMBO Rep. 2002;3(12):1146-1151.
doi pubmed - Ascenzi P, di Masi A, Leboffe L, Fiocchetti M, Nuzzo MT, Brunori M, Marino M. Neuroglobin: From structure to function in health and disease. Mol Aspects Med. 2016;52:1-48.
doi pubmed - Raychaudhuri S, Skommer J, Henty K, Birch N, Brittain T. Neuroglobin protects nerve cells from apoptosis by inhibiting the intrinsic pathway of cell death. Apoptosis. 2010;15(4):401-411.
doi pubmed - Yu Z, Liu N, Liu J, Yang K, Wang X. Neuroglobin, a novel target for endogenous neuroprotection against stroke and neurodegenerative disorders. Int J Mol Sci. 2012;13(6):6995-7014.
doi pubmed - Ciccone L, Nencetti S, Socci S, Orlandini E. Neuroglobin and neuroprotection: the role of natural and synthetic compounds in neuroglobin pharmacological induction. Neural Regen Res. 2021;16(12):2353-2358.
doi pubmed - Rinaldi C, Wood MJA. Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat Rev Neurol. 2018;14(1):9-21.
doi pubmed - Marxreiter F, Stemick J, Kohl Z. Huntingtin lowering strategies. Int J Mol Sci. 2020;21(6):2146.
doi pubmed - Pan L, Feigin A. Huntington's disease: new frontiers in therapeutics. Curr Neurol Neurosci Rep. 2021;21(3):10.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Neurology Research is published by Elmer Press Inc.