

| Journal of Neurology Research, ISSN 1923-2845 print, 1923-2853 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Neurol Res and Elmer Press Inc |
| Journal website http://www.neurores.org |
Original Article
Volume 6, Number 4, August 2016, pages 57-64
The Criteria for Diagnosing Amyotrophic Lateral Sclerosis May Be Unsuitable for Clinical Use
Syuichi Tetsuka
Department of Neurology, Hospital of International University of Health and Welfare, 537-3, Iguchi, Nasushiobara, Tochigi 329-2763, Japan
Manuscript accepted for publication July 20, 2016
Short title: Diagnosing Amyotrophic Lateral Sclerosis
doi: http://dx.doi.org/10.14740/jnr383w
| Abstract | ▴Top |
Background: The existing diagnostic criteria for definite, probable and possible amyotrophic lateral sclerosis (ALS) require at least one upper motor neuron (UMN) sign. However, we think some cases of ALS or motor neuron disease may be not able to fulfill the required criteria. We investigated whether the clinical presentation and course of patients with ALS or motor neuron disease fulfill the required criteria or not.
Methods: In total, 296 patients who were diagnosed with ALS or motor neuron disease in the institute in the past 15 years were retrospectively investigated and analyzed.
Results: In total, 108 patients (36.5%) who exhibited lower motor neuron (LMN) signs but not UMN signs at the early stage of the disease were diagnosed with ALS or motor neuron disease. Sixty-four of these 108 patients (59.2%) who developed respiratory failure and swallowing difficulty or UMN signs during the 5-year follow-up period were diagnosed with clinical ALS. No significant difference in the survival probability was observed between patients who exhibited both UMN and LMN signs and patients who exhibited only LMN signs.
Conclusion: These findings suggest that some of motor neuron disease cases who only exhibit LMN signs can be diagnosed as ALS. I suggest the revised El Escorial Criteria for the Diagnosis of ALS may be unsuitable for clinical use.
Keywords: Amyotrophic lateral sclerosis; Adult-onset sporadic SMA; Criteria; Diagnosis; Progressive muscular atrophy; Spinal muscular atrophy
| Introduction | ▴Top |
Amyotrophic lateral sclerosis (ALS) is the most common and most severe form of motor neuron disease, eventually leading to death due to respiratory insufficiency within 3 - 5 years. ALS is characterized by the degeneration of lower motor neurons (LMNs) in the anterior horn of the spinal cord or in the brainstem and of upper motor neurons (UMNs) in the motor cortex of the brain [1].
ALS phenotypes can be classified in many ways, and there is our preferred clinical classification [2] (Table 1). Progressive muscular atrophy (PMA) that is one of ALS types is clinically characterized solely by signs of LMN dysfunction. PMA patients who at any time later during follow-up develop UMN signs are then considered to have LMN-onset ALS [3]. However, the terminology called as PMA is not well recognized among neurological clinicians who actually treat these patients at present. Spinal muscular atrophy (SMA) is a hereditary neurodegenerative disease that mainly affects LMNs. It is divided into three types according to the age of onset and severity: type I (Werdnig-Hoffmann disease), type II (the intermediate form), and type III (Kugelberg-Welander disease) [4]. Although some SMA patients have an SMN gene mutation similar to that seen in childhood-onset types I-III [5], these cases are rare [4].
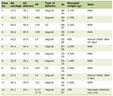 Click to view | Table 1. ALS Phenotypes as Recognized by Clinicians |
The broad sense of SMA includes adult-onset SMA and childhood-onset SMA. Therefore, it is clinically difficult to distinguish the broad definition of PMA from adult-onset sporadic SMA [3], which is also known as type IV SMA. Adult-onset SMA is characterized as a sporadic disease in most cases, with onset in adulthood and old age and slow disease progression. Some cases exhibit muscular atrophy, muscle weakness, muscle fiber fasciculation, and decreased tendon reflex at distal upper extremities at the early stage of the disease, and these signs are gradually expanded to the whole body with motor dysfunction. In other cases, the disease onset is characterized by proximal muscle atrophy, particularly shoulder girdle muscle atrophy. Unlike ALS, the prognosis for adult-onset sporadic SMA is not death from the disease [6]. Although differential diagnosis of ALS with pure LMN involvement from another motor neuron disease, spinal bulbar muscular atrophy (Kennedy-Alter-Sung syndrome), is important, this polyglutamine disease can be diagnosed by genetic testing at present [7].
Whether PMA is an independent disease or a subtype of ALS remains unclear at the early stage of the disease. It is also difficult to distinguish adult-onset sporadic SMA from motor neuropathy. Furthermore, existing diagnostic criteria for definite, probable and possible ALS require at least one UMN sign [3, 8]. However, in daily clinical consultations, many neurologists frequently encounter problems diagnosing cases of motor neuron disease using this diagnostic criterion. Therefore, in the present study, we investigated the clinical presentations and courses of PMA patients who only exhibited LMN signs and whether it is possible to diagnose as ALS disease using the revised El Escorial Criteria for the Diagnosis of ALS.
| Materials and Methods | ▴Top |
Patient data
In total, 296 patients who were diagnosed with ALS or motor neuron disease by a neurologist during the past 15 years in Jichi Medical University Hospital in Japan were retrospectively investigated. To diagnose ALS or motor neuron disease, not only clinical examination but also electromyography was conducted. Furthermore, a nerve conduction study, magnetic resonance imaging, transcranial magnetic stimulation and examination of cerebrospinal fluid were performed to distinguish other neuronal diseases such as multifocal motor neuropathy, myelopathy, and inclusion body myositis which were confirmed by muscular biopsy. In addition, patients with spinal bulbar muscular atrophy (Kennedy-Alter-Sung syndrome) who had only LMN involvement were excluded according to genetic diagnosis. The final study population included 158 male patients with a mean age of 61.8 years and 138 female patients with a mean age of 62.2 years.
Retrospective observation
In this retrospective review, both the outcomes and clinical manifestations of these patients were evaluated. Survival time, an endpoint in this study, was defined as the number of months from symptom onset until death from any cause or of tracheostomy for institution of permanent mechanical ventilation and incidence of non-invasive ventilation. The patient characteristics entered in univariate analyses were the age at onset, sex, and bulbar symptoms. Moreover, the onset of UMN signs in the patients who had only LMN signs was entered during follow-up periods. In addition, the death, respiratory failure, gastrostomy or swallowing disturbance, and developed UMN signs are the four measurements used as features for the diagnosis of clinical ALS. The retrospective analysis was ethically admitted without health risk by the Research Committee of CNS Degenerative Diseases of Japan.
Statistical analysis
For general statistical analyses, we used SPSS v.11.0.1 program. χ2 tests for independent testing were applied to a two-by-two contingency table with sex and bulbar symptoms. Student’s t-tests were applied to test differences in the mean age at onset. The effects of prognostic factors on survival were assessed using the Kaplan-Meier life-table method for all 296 patients according to the presentation of UMN and LMN signs or only LMN signs. The log-rank test was used to assess the equality of the outcome functions. All tests were two-tailed, and significance was set at P < 0.05.
| Results | ▴Top |
Percentages of patients with motor neuron diseases according to motor neuron symptoms and characteristics
Of 296 patients that were diagnosed as motor neuron diseases, 188 patients (63.5%) who had UMN and LMN signs at the time of admission were diagnosed with ALS. In 108 patients (36.5%) who exhibited only LMN signs, ALS could not be confirmed (Fig. 1A). Even magnetic resonance imaging and transcranial magnetic stimulation could not confirm UMN signs in these patients. The mean age at onset of motor neuron dysfunction was 61.8 ± 11.2 years in patients with UMN and LMN signs and 62.3 ± 10.7 years in those with only LMN signs, with no significant difference between these ages (t-test, P = 0.76, Table 2). The percentages of patients of either sex also were not significantly different between the groups (χ2 test, P = 0.93, Table 2). On the other hand, a significantly higher number of patients had a bulbar symptom and UMN and LMN signs than LMN signs only (χ2 test, P < 0.05, Table 2). We found that there were more progressive bulbar palsy-type ALS cases among the patients exhibiting both UMN and LMN signs.
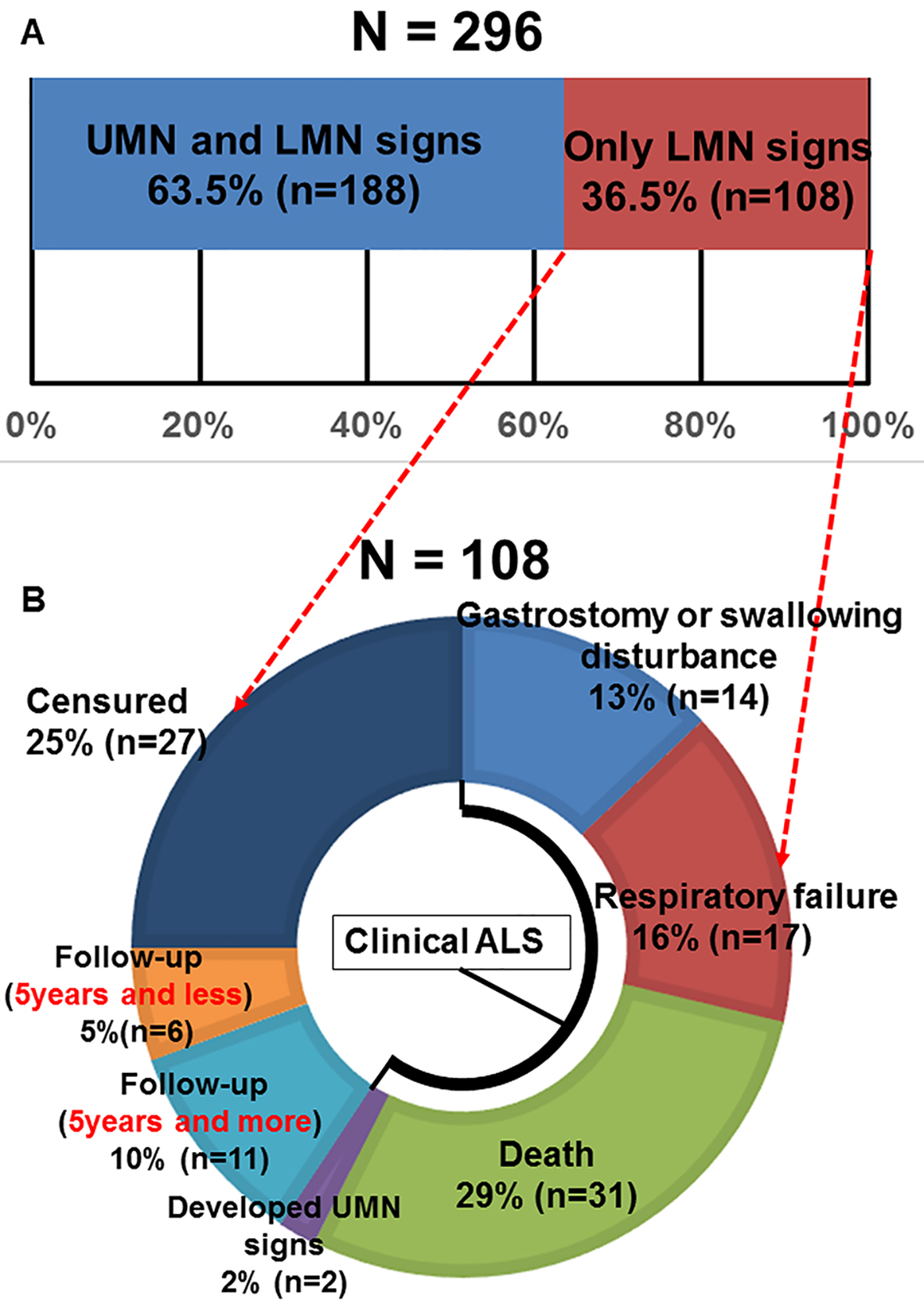 Click for large image | Figure 1. (A) Motor neuron disease patient’s percentages according to motor neuron signs (UMN: upper motor neuron; LMN: lower motor neuron). (B) The outcome of 108 cases whose definite, probable and possible ALS diagnosis was unable to be confirmed due to only LMN signs during follow-up periods. |
Click to view | Table 2. Baseline Characteristics of Patients With Those Motor Neuron Disease |
Outcomes of 108 patients with only LMN dysfunction
Figure 1B shows the outcomes of 108 patients whose ALS diagnosis could not be confirmed because of the presentation of only LMN signs. Thirteen patients (14%) had gastrostomy or deteriorated swallowing disturbance. Seventeen patients (16%) developed respiratory failure. Thirty-one patients (29%) died. Two patients (2%) developed UMN signs during 5-year follow-up. These results indicate that a total of 64 patients (59.2%), including the patients listed above, were diagnosed with ALS during the 5-year follow-up period on the basis of their poor prognosis and development of UMN signs, which is unlike adult-onset sporadic SMA. In summary, the death, respiratory failure, gastrostomy or swallowing disturbance, and developed UMN signs are the four measurements used as clinical features for the diagnosis of clinical ALS (Fig. 1B). Eleven patients (10%) continued to exhibit only LMN signs for 5 years and more. Six patients continued to exhibit only LMN signs for less than 5 years during follow-up, and 27 patients (25%) did not complete follow-up examination because of moving out to other institutes (i.e., censured).
Survival probability in both groups according to motor neuron symptoms (both UMN and LMN or only LMN)
The Kaplan-Meier survival curves of 188 ALS patients who exhibited both UMN and LMN signs and 108 patients who exhibited only LMN signs are shown for all 296 patients in Figure 2A. There was a statistically significant difference between these survival curves (log-rank test, P < 0.05). Patients who only exhibited LMN signs had a higher survival probability than those who exhibited both UMN and LMN signs, and several patients survived for a long period. However, the time from onset to death was limited to 5 years by examination, and the Kaplan-Meier survival curves of the 260 patients (173 with both UMN and LMN signs and 87 with only LMN signs) with vs. without UMN signs did not reveal any differences (log-rank test, P = 0.06, Fig. 2B). As a result, when the time from onset to death was limited to 5 years, there was a no significant difference in the survival curves (Fig. 2B) unlike the period without limitation (Fig. 2A).
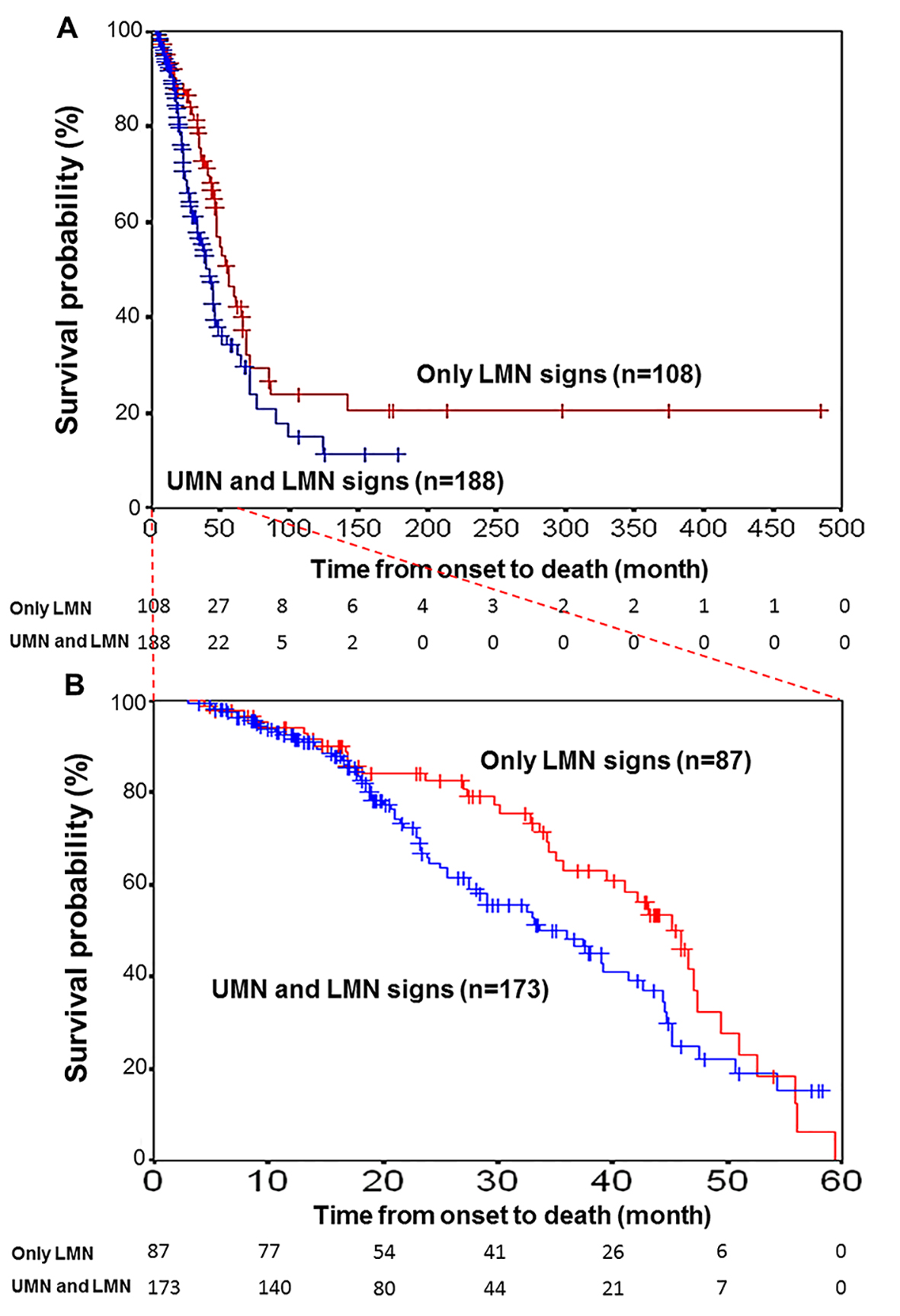 Click for large image | Figure 2. Kaplan-Meier survival curves for patients with motor neuron disease. (A) Kaplan-Meier survival curves for motor neuron disease patients (N = 188) with UMN and LMN signs (blue line) versus those (N = 108) with only LMN signs (red line) for all 296 patients according to the motor neuron symptoms status. Vertical bars indicate censured data. A significant difference was found between the two survival curves (log-rank test, P < 0.05). (B) Time from onset to death was limited to 5 years by examination. The curves of 260 patients (173 with both UMN and LMN signs and 87 with only LMN signs) with vs. without UMN signs did not reveal any difference (log-rank test, P = 0.06). |
Disease duration in patients who had only LMN dysfunction and were not diagnosed with ALS
Eleven patients who exhibited only LMN signs for 5 years and more included four males and seven females. The average age at disease onset was 57.2 years in male patients and 50.4 years in female patients. The primary symptoms were unilateral muscle weakness of the upper extremities in seven patients, unilateral muscle weakness of the lower extremities in three patients, and bilateral muscle weakness in the lower extremities in one patient. Figure 3 shows the duration of disease in patients with only LMN signs for 5 years and more (5 - 10 years in five patients, 10 - 15 years in two patients, 15 - 20 years in one patient, and more than 20 years in three patients). Some patients exhibited only LMN signs over lengthy periods. The abilities of these 11 patients to complete activities of daily living were as follows: seven patients were ambulatory, four patients were not ambulatory, four patients were self-sufficient in daily life, six patients required assistance in daily life, and one patient had swallowing difficulty and required assistance in daily life. The longest retrospective history of a patient for ALS patients was approximately 480 months (Fig. 2A).
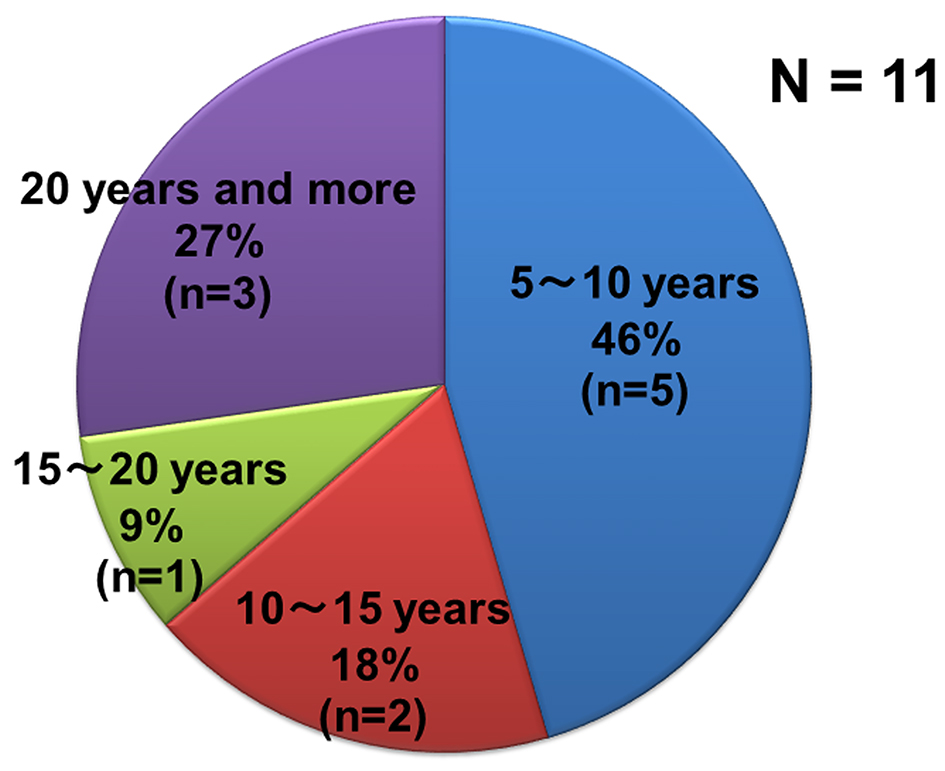 Click for large image | Figure 3. Percentage of disease durations of patients with only LMN signs and who have been not diagnosed as ALS. |
Example cases of motor neuron disease having only LMN signs
Case 1 (had only LMN signs for 5 years and more): 53-year-old female patient; disease onset at 38 years old; muscle weakness in the left lower extremities. Clinical course: the patient was hospitalized at our institute when she was 44 years old because of muscle weakness of the extremities (manual muscle testing: deltoid 3/3, wrist 5/5, iliopsoas 3/3, and tibialis anterior 1/1) and a decrease in deep tendon reflexes. Electromyography showed active neurogenic change in the extremity muscles. The muscle weakness gradually progressed. Recent clinical condition: manual muscle testing: deltoid 1/1, wrist 4/4, iliopsoas 1/1, and tibialis anterior 0/0. The patient was unable to walk. At 14 years after disease onset, UMN signs had not developed. She died because of right putaminal hemorrhage in 53 years old. Autopsies were performed. Histological examination showed not only atrophy of the anterior horns of the spinal cord, but also the extensive dropout of anterior horn cells in lumbar cord and cervical spinal cord (Fig. 4).
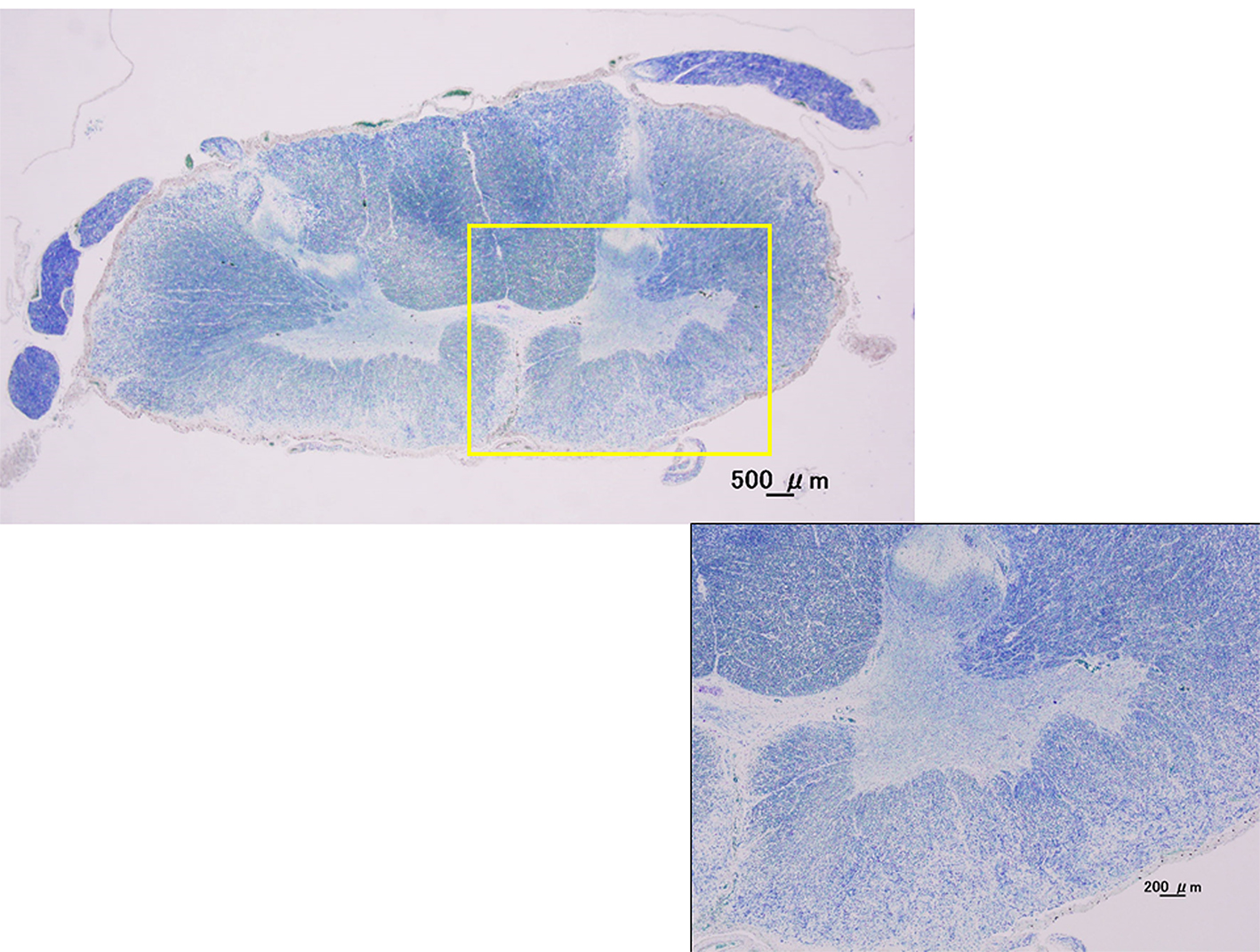 Click for large image | Figure 4. Kluver-Barrera stain. Cervical spinal cord (C7). Histological examination showed not only atrophy of the anterior horns of the spinal cord, but also the extensive dropout of anterior horn cells in cervical spinal cord with the pure LMN cases by autopsies. |
Case 2 (had only LMN signs for 5 years and more): 65-year-old female patient; disease onset at 41 years old; muscle weakness in both lower extremities. Clinical course: this patient had a family history (older sister, younger brother, and cousin) of similar disease symptoms. The patient exhibited muscle weakness of the extremities (manual muscle testing: deltoid 4/4, wrist 3/4, iliopsoas 3/3, and tibialis anterior 4/4) and lost DTRs at 61 years of age. Electromyography showed inactive neurogenic change in the extremity muscles. The muscle weakness gradually progressed. Recent clinical condition: manual muscle testing: deltoid 3/3, wrist 3/3, iliopsoas 3/3, and tibialis anterior 4/4. Compared with 4 years previously, the symptoms progressed very slowly. The patient was still able to walk with assistance 24 years after disease onset.
| Discussion | ▴Top |
Cases of adult-onset motor neuron diseases that exhibit only LMN signs can be diagnosed as PMA, although the terminology is not well recognized among neurological clinicians. According to the ALS diagnostic criteria, definite, probable and possible ALS diagnosis requires at least one UMN sign [8]. Therefore, it is difficult to diagnosis these patients as ALS. Adult-onset sporadic SMA (type IV): 1) is sporadic in many cases; 2) begins in adulthood or old age; and 3) shows slow disease progression that results in a relatively favorable prognosis. It has not yet been concluded whether it is an independent disease or subtype of ALS. In addition, it is difficult to distinguish this disease from motor neuropathy in many cases. In the present study of 296 motor neuron disease patients, we found that 108 patients (36.5%) did not exhibit UMN signs and exhibited only LMN signs during early stage disease. However, 64 of these patients (59.2%) developed respiratory failure and swallowing disturbance or UMN signs during 5-year follow-up, resulting in the diagnosis of ALS rather than adult-onset sporadic SMA at later stages of the disease, using the four measurements as clinical features for the diagnosis of clinical ALS (gastrostomy or swallowing disturbance, and developed UMN signs) which are based on evidence that ALS prognosis is within 3 - 5 years. This was because their prognosis was poor and they developed UMN signs during follow-up, which are not observed in adult-onset sporadic SMA. Although patients with adult-onset sporadic SMA tended to live longer than those with ALS, shorter survival in adult-onset sporadic SMA is associated with the same risk factors that predict poor survival in ALS [1].
Some of the results of this study are well-known as the longer duration of PMA than those of classical ALS. In the present study, patients who exhibited only LMN signs also had a higher survival probability than those who exhibited UMN and LMN signs, and several patients survived for a long period after disease onset. The results indicate that the group of 108 patients who exhibited only LMN signs may have included some patients with adult-onset sporadic SMA who had the abovementioned clinical characteristics. In addition, this study indicated that a few adult-onset sporadic SMA patients with one affected body region, as observed in the flail arm and leg syndromes, should be considered distinct from a motor neuron disease like ALS because symptom progression in these syndromes is much slower than in ALS [9, 10], and we indicated these results even in case examples. In addition, because it is important to make the differential diagnosis of ALS from adult-onset sporadic SMA, further their genetic background (SMN1 and SMN2) analysis is required for the presence of adult-onset sporadic about these 11 cases (Fig. 3) [5]. This is an issue in the future. On the other hand, some studies reported that some of their PMA patients had an “ALS-like” disease course with aggressive disease progression and early death, although they studied fewer number of patients compared with our study [11, 12]. Survival probability in both groups according to motor neuron signs (both UMN and LMN or only LMN) in this retrospective study was similar to these studies, when the time from onset to death was limited to 5 years by examination. However, the medical positioning between ALS, PMA and adult-onset sporadic SMA is unclear at present, when neurological clinicians actually treat these patients. Therefore, we indicate the positioning in Figure 5 on the basis of this study. Pathologic studies have shown UMN involvement which Betz cells had presumably disappeared, and absence of astrocytosis of the V layer in the primary motor cortex and pyramidal tract degeneration in patients with only LMN signs who were clinically diagnosed while alive with PMA [13, 14]. We also confirmed the atrophy of the anterior horns of the spinal cord and the extensive dropout of anterior horn cells in lumbar cord and cervical spinal cord in the case1 with the pure LMN cases by autopsies. It is generally accepted that the time from ALS onset to death due to respiratory insufficiency is 3 - 5 years. In addition, we found no difference in survival probability between patients who exhibited both UMN and LMN signs and those who exhibited only LMN signs within 5 years; however, a significant difference was detected during the 40-year follow-up. Patients who only exhibited LMN signs had a higher survival probability than those who exhibited both UMN and LMN signs, and several patients survived for long periods. These results indicate that the group of patients who exhibited only LMN signs may have been influenced by including patients with adult-onset sporadic SMA. Therefore, patients who only exhibited LMN signs and died within 5 years were diagnosed with ALS retrospectively. Thus, the 5-year rule may be applicable for diagnosing ALS. We aimed to distinguish ALS from adult-onset sporadic SMA according to the disease duration of at least 5 years.
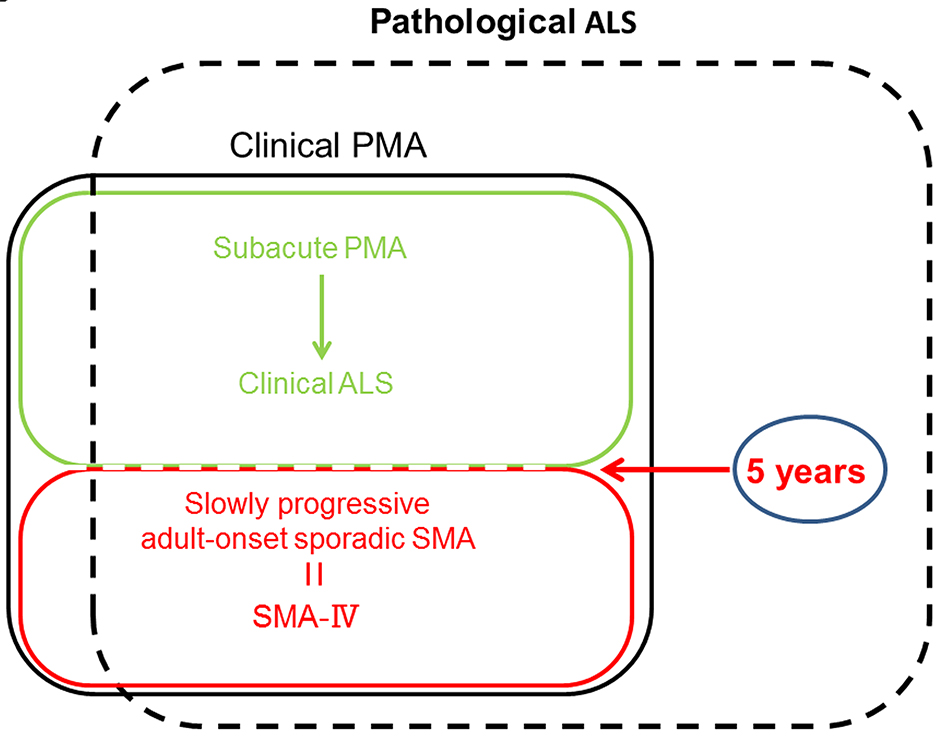 Click for large image | Figure 5. The medical positioning between ALS, PMA and adult-onset sporadic SMA. PMA: progressive muscular atrophy; SMA: spinal muscular atrophy. |
However, the numbers of adult-onset sporadic SMA among groups of patients with motor neuron diseases are small. In most PMA cases, disease progression during the early stage of the disease is relentlessly progressive, eventually leading to death because of respiratory insufficiency within a few years. Therefore, PMA should be considered as a form of ALS. These results suggest that diagnostic confirmation of motor neuron disease according to clinical symptoms at the early stage of the disease is difficult, and patient follow-up is critical. In addition, our results indicate that some ALS patients exhibit only LMN signs during the early stages of the disease. Because existing diagnostic criteria for definite, probable and possible ALS require at least one UMN sign, it may be necessary to revise these criteria to make them more applicable in clinical use. Traynor et al advocated that the El Escorial [15] are excessively restrictive, and levels of diagnostic certainty cannot be used as prognostic indicators [16]. Thereafter, the revised El Escorial Criteria for the Diagnosis of ALS were established in 2000 [8], moreover the Awaji criteria which have been proven to be more sensitive in the diagnosis of ALS by making much account of electromyography studies’ outcome were advocated in 2008 [17]. But it was not basically different from the revised El Escorial Criteria with regard to the restrictiveness. Utility of the revised El Escorial Criteria is immense for clinical research, pharmacological trials and exchange information among research centers. In addition, the El Escorial clinical criteria for the diagnosis of ALS are considered very reliable, with virtually absent risk of false-positive diagnosis [18, 19]. However, this study indicates that the clinical presentation and course of some ALS patients have not adhered to the criteria in the point of view of neurological clinicians who actually treat these patients. European EFNS Task Force on Management of Amyotrophic Lateral Sclerosis in 2005 also did not recommend that patients are told they have definite, probable and possible ALS in practice. The clinician must decide, on the balance of probability, whether or not the patient has ALS, even in the absence of unequivocal UMN and LMN signs [20]. The neurological clinician would discuss with those patients about these very percentages, and counsel that ALS might be in their future. As neurological clinician, we think that it is ideal consultation. However, according to the international criteria for the diagnosis of ALS that are too restrictive, we speculate that some countries create their guideline for ALS like Japan. As the results, I just want to advocate that according to the international criteria for the diagnosis of ALS that are too restrictive, some ALS patients that do not fulfill the required criteria for the diagnosis of ALS might not be able to receive medical welfare aid with regard to intractable neurological diseases like Japan. I think that some neurological clinicians following ALS patients frequently encounter in daily clinical consultation.
Conclusion
In conclusion, some cases of motor neuron disease in which patients exhibited only LMN signs can be diagnosed as having ALS by follow-up of their clinical course. In addition, some ALS patients suffer a disadvantage because of the international criteria for the diagnosis of ALS that are too restrictive. In addition, the 5-year rule may be reasonable for clinically diagnosing ALS. Therefore, I propose that the revised El Escorial Criteria for the Diagnosis of ALS may be unsuitable for clinical use.
Acknowledgments
I thank Dr. Mitsuya Morita (Jichi Medical University) for helping the collection of patients’ data when I was enrolled in Jichi Medical University. This work was supported by Grants-in-Aid from the Research Committee of CNS Degenerative Diseases, the Ministry of Health, Labour and Welfare of Japan.
Conflicts of Interest
The authors have no conflicts of interests to declare.
| References | ▴Top |
- Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, Burrell JR, et al. Amyotrophic lateral sclerosis. Lancet. 2011;377(9769):942-955.
doi - Al-Chalabi A, Hardiman O. The epidemiology of ALS: a conspiracy of genes, environment and time. Nat Rev Neurol. 2013;9(11):617-628.
doi pubmed - Kim WK, Liu X, Sandner J, Pasmantier M, Andrews J, Rowland LP, Mitsumoto H. Study of 962 patients indicates progressive muscular atrophy is a form of ALS. Neurology. 2009;73(20):1686-1692.
doi pubmed - D'Amico A, Mercuri E, Tiziano FD, Bertini E. Spinal muscular atrophy. Orphanet J Rare Dis. 2011;6:71.
doi pubmed - Feldkotter M, Schwarzer V, Wirth R, Wienker TF, Wirth B. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet. 2002;70(2):358-368.
doi pubmed - Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371(9630):2120-2133.
doi - Katsuno M, Banno H, Suzuki K, Takeuchi Y, Kawashima M, Tanaka F, Adachi H, et al. Molecular genetics and biomarkers of polyglutamine diseases. Curr Mol Med. 2008;8(3):221-234.
doi pubmed - Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1(5):293-299.
doi pubmed - Vucic S, Kiernan MC. Abnormalities in cortical and peripheral excitability in flail arm variant amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2007;78(8):849-852.
doi pubmed - Wijesekera LC, Mathers S, Talman P, Galtrey C, Parkinson MH, Ganesalingam J, Willey E, et al. Natural history and clinical features of the flail arm and flail leg ALS variants. Neurology. 2009;72(12):1087-1094.
doi pubmed - Van den Berg-Vos RM, Visser J, Kalmijn S, Fischer K, de Visser M, de Jong V, de Haan RJ, et al. A long-term prospective study of the natural course of sporadic adult-onset lower motor neuron syndromes. Arch Neurol. 2009;66(6):751-757.
doi pubmed - Visser J, van den Berg-Vos RM, Franssen H, van den Berg LH, Wokke JH, de Jong JM, Holman R, et al. Disease course and prognostic factors of progressive muscular atrophy. Arch Neurol. 2007;64(4):522-528.
doi pubmed - Ince PG, Evans J, Knopp M, Forster G, Hamdalla HH, Wharton SB, Shaw PJ. Corticospinal tract degeneration in the progressive muscular atrophy variant of ALS. Neurology. 2003;60(8):1252-1258.
doi pubmed - Tsuchiya K, Sano M, Shiotsu H, Akiyama H, Watabiki S, Taki K, Kondo H, et al. Sporadic amyotrophic lateral sclerosis of long duration mimicking spinal progressive muscular atrophy exists: additional autopsy case with a clinical course of 19 years. Neuropathology. 2004;24(3):228-235.
doi pubmed - Brooks BR. El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial "Clinical limits of amyotrophic lateral sclerosis" workshop contributors. J Neurol Sci. 1994;124(Suppl):96-107.
doi - Traynor BJ, Codd MB, Corr B, Forde C, Frost E, Hardiman OM. Clinical features of amyotrophic lateral sclerosis according to the El Escorial and Airlie House diagnostic criteria: A population-based study. Arch Neurol. 2000;57(8):1171-1176.
doi pubmed - de Carvalho M, Dengler R, Eisen A, England JD, Kaji R, Kimura J, Mills K, et al. Electrodiagnostic criteria for diagnosis of ALS. Clin Neurophysiol. 2008;119(3):497-503.
doi pubmed - Kurian KM, Forbes RB, Colville S, Swingler RJ. Cause of death and clinical grading criteria in a cohort of amyotrophic lateral sclerosis cases undergoing autopsy from the Scottish Motor Neurone Disease Register. J Neurol Neurosurg Psychiatry. 2009;80(1):84-87.
doi pubmed - Makki AA, Benatar M. The electromyographic diagnosis of amyotrophic lateral sclerosis: does the evidence support the El Escorial criteria? Muscle Nerve. 2007;35(5):614-619.
doi pubmed - Andersen PM, Borasio GD, Dengler R, Hardiman O, Kollewe K, Leigh PN, Pradat PF, et al. EFNS task force on management of amyotrophic lateral sclerosis: guidelines for diagnosing and clinical care of patients and relatives. Eur J Neurol. 2005;12(12):921-938.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Neurology Research is published by Elmer Press Inc.