

| Journal of Neurology Research, ISSN 1923-2845 print, 1923-2853 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Neurol Res and Elmer Press Inc |
| Journal website http://www.neurores.org |
Original Article
Volume 3, Number 3-4, August 2013, pages 122-129
Autistic Symptoms in Late Diagnosed Phenylketonuric Children in Upper Egypt
Khaled Saada, d, Emad Hammada, Ahmed A. Abdel-rahmanb, Karema M. Sobhyc
aDepartment of Pediatrics, Assiut University, Assiut 71516, Egypt
bDepartment of Neuropsychiatry, Assiut University, Assiut 71516, Egypt
cDepartment of Public Health, Assiut University, Assiut 71516, Egypt
dCorresponding author: Khaled Saad, Department of Pediatrics, Faculty of Medicine, University of Assiut, Assiut 71516, Egypt
Manuscript accepted for publication July 24, 2013
Short title: Autistic Symptoms
doi: https://doi.org/10.4021/jnr221w
| Abstract | ▴Top |
Background: Phenylketonuria (PKU) is the most common metabolic cause of mental retardation. Increased concentrations of Phe in PKU have a neurotoxic effect, contributing to the structural brain damage, severe mental retardation, and psychiatric disturbances. Co-occurrence of autism and PKU has been described in several studies. The toxic levels of Phe appear to play a significant role in the development of autism symptomatology. Our objective was to assess autistic features in a cohort of late diagnosed PKU children.
Methods: Thirty-two PKU pediatric patients diagnosed after the age of one year and followed up at Pediatric University Hospital, Assiut University, Egypt. DSM-IV TR and CARS were used for diagnosis and classification of autism cases. FSIQ and MRI brain were done for all cases. EEG was done for cases with convulsions.
Results: Autism was present in 8 (25%) cases; 3 were females and 5 were males, 2 had severe autism and 6 had mild/moderate autism, 5 patients had classic PKU and 3 had moderate PKU, 3 cases had mild mental retardation and five have moderate mental retardation.
Conclusion: Delay in the diagnosis and management of PKU leads to very high incidence of intellectual disabilities and autistic features in these patients.
Keywords: Phenylketonuria; Autism; EEG
| Introduction | ▴Top |
Phenylketonuria (PKU) is the most prevalent disorder caused by an inborn error in amino acid metabolism. It is the most common metabolic cause of mental retardation [1]. The disorder is characterized by deficiency in the hepatic enzyme phenylalanine hydroxylase (PAH) or its cofactor tetrahydrobiopterin (BH4). Phenylalanine (Phe) cannot be converted to tyrosine (Tyr); this results in the harmful buildup of Phe in the body and a deficiency of Tyr, a precursor to the neurotransmitters dopamine and norepinephrine in the central nervous system. Phenotypes can vary from a mild increase in blood Phe concentrations to a severe classic phenotype with pronounced hyperphenylalaninaemia. Increased concentrations of Phe have a neurotoxic effect, contributing to the structural brain damage, severe mental retardation, and psychiatric disturbances [1-4]. The incidence of PKU in Caucasian populations is between 1 in 10,000 and 1 in 15,000 people, with higher incidence in Turkey (1 in 2,600) and Arabic populations (1 in 6,000). It has been suggested that this higher incidence of PKU is due to the high prevalence of consanguinity [1, 5]. Early identification of PKU through newborn screening and early initiation of treatment with a Phe-restricted diet have prevented the most severe complications of this disorder. However, psychiatric disorders, behavioral problems, and cognitive deficits commonly occur [6]. There are three mechanisms through which elevated Phe indirectly affects brain function, including myelin abnormalities, disruption of amino acid transport across the blood-brain barrier, and reductions in neurotransmitters. Magnetic resonance imaging (MRI) studies document an association between high Phe levels and reduced myelin. Although the mechanism by which Phe affects myelin is not clear, myelination is delayed in untreated or poorly treated children with PKU, and dysmyelination occurs in adults who discontinue the Phe-restricted diet [7, 8].
Autism is a behaviorally defined syndrome beginning before 3 years of age that is characterized by pervasive deficits in social interaction, impairment in verbal and nonverbal communication, and stereotyped patterns of interests and activities. Autism spectrum disorder is present in 1 in 150 to 166 individuals, with 3 to 6 more boys affected than girls. Several neuropathologic changes, including abnormalities in the hippocampus, amygdala, cerebellum, and cerebral cortex, have been described [9, 10]. Co-occurrence of autism and PKU has been described in several studies [11-13], although the strength of the association is inconsistent and difficult to determine as a result of significant improvements in early identification and treatment. The exact percentage of individuals with phenylketonuria who display autistic features is difficult to determine, especially since the introduction of treatment. Studies found an autism frequency ranging from 2.7% to 5.7% in individuals with PKU [14, 15]. The association between autism and PKU has important implications for understanding possible underlying factors in the development of autism. In the case of PKU, the toxic levels of Phe appear to play a significant role in the development of autism symptomatology [14]. Biogenic amines have been shown to facilitate formation of synapses in the developing brain. The accumulation of Phe in patients with PKU with lack of biogenic amines in the brain is associated with the presence of mental retardation and autism [16].
The current study was conducted to assess autistic features in a cohort of late diagnosed PKU children (namely first diagnosed with phenylketonuria after at least one year of age) in Assiut, Upper Egypt.
| Material and Methods | ▴Top |
Thirty-two PKU pediatric patients diagnosed late after the age of one year and followed up at Pediatric University Hospital, Assiut University, Egypt, from Jan 2004 until Jan 2012.
Inclusion criteria: 1), PKU Patients diagnosed 1 year or more after birth; 2), No evidence of any other systemic or metabolic diseases; 3), No history of significant head injury or neurological disease.
Exclusion criteria: early and continuously treated PKU patients.
Informed written consent was obtained from each caregiver. The study was approved by the Ethical Committee of Assiut University, Assiut, Egypt.
The study was conducted over a 1.5 year period. Eligible subjects were evaluated according to the following.
Detailed history with carefully gathered information by structured parent interview, paying special attention to family history of consanguinity, similar conditions of PKU in the family, time of onset of PKU symptoms, autistic symptoms, social/interpersonal activities, self care, pattern of any cognitive decline, behavioral and learning disorders, delayed motor milestones, mental retardation, seizures and prenatal and perinatal history.
General and meticulous neurological examination:(consisting of anthropometric measurements, testing limb tone and strength, tendon reflexes, tremors, and tests for cerebellar function).
Full Scale IQ (FSIQ) test: by applying Stanford Binet Intelligence Scale, Fourth Edition; the test is valid by using factorial validity, criterion validity and also some studies operate in the culture of Egypt that sub testes of Stanford-Binet (4th edition) [17]. For a standardized definition of mental retardation, DSM IV TR requires an IQ of approximately 70 or below on an individually administered IQ test with concurrent deficits or impairments in present adaptive functioning (namely, the person’s effectiveness in meeting the standards expected for his or her age by his or her cultural group) in at least two of the following areas: communication, self-care, home living, social/ interpersonal skills, use of community resources, self-direction, functional academic skills, work, leisure, health, and safety provided that onset is before the age of 18 years. Individuals with IQ scores of 50 - 70 are classified as having mild mental retardation; those with IQ level of 35 - 50 have moderate mental retardation, those with IQ level ranging from 20 to 35 have severe mental retardation and finally subjects with an IQ level below 20 are classified as having profound mental retardation. IQ scores of 70 - 90 are often classified as borderline and scores of 90 or higher are considered normal [18].
Autistic disorder was diagnosed according to DSM-IV TR diagnostic criteria [19], structured interviews of at least one hour each both with the parents and the child were performed, in a room equipped with play material appropriate for age level. Later on, another two hours-session was conducted for classification of autism cases by using the Childhood Autism Rating Scale (CARS) [20]. CARS is a well-established scale for the screening and diagnosis of childhood autism with complete agreement between DSM-IV TR diagnostic criteria and CARS [21]. The scale assesses behavior in 14 domains that are generally affected by severe problems in autism, plus one general category of impressions of autism, with the aim of identifying children with autism, as differentiated from the other developmental disorders. The 15 items in the scale are: relating to people; imitative behavior; emotional response; body use; object use; adaptation to change; visual response; listening response; perceptive response; fear or anxiety; verbal communication; non-verbal communication; activity level; level; consistency of intellective relations and general impressions. The examiner assigned a score of 1 to 4 for each item: 1 indicates behavior appropriate for age level, while 4 indicates severe deviance with respect to normal behavior for age level. The scores for the single items are added together into a total score, which classifies the child as not autistic (below 30), mild or moderately autistic (30 - 36.5) or severely autistic (above 36.5) [20, 21].
Phenylalanine Levels: Concurrent levels for all cases with PKU (taken directly after completion of psychological assessment) were included. The results were compared with previous recent Phe levels to assess the representativeness of the concurrent level. PKU is classified into three categories based on severity at diagnosis: classic PKU (plasma Phe > 1,200 µmol/L), moderate PKU (plasma Phe > 600 - 1,200 µmol/L), or mild PKU (plasma Phe 360 - 600 µmol/L) [22].
EEG was done for cases with convulsions, MRI brain was done for all cases.
Statistical analysis
The statistical study was performed with the SPSS program (version 19.0). Pearson chi-square test was applied to search for association between categorical variables. ANOVA correction was used to compare quantitative variables when the number of data was higher than 30, the distribution was normal and the variances were not significantly different (Levene test). Otherwise Pearson test was used for correlations between quantitative data. Statistical significance was accepted for P < 0.05.
| Results | ▴Top |
Thirty-two PKU pediatric patients were enrolled in this study after obtaining the parents informed consent. Table 1 shows demographic data and classification of patients. The age at diagnosis ranged from 3 years to 8 years (5.47y± 1.45 y). Late diagnosis of PKU in studied patients is attributed to the lack of screening program for PKU in Egypt. The study included 22 males (68.80%) and ten females (31.20%), 20 patients (62.50%) had classic PKU; 16 patients (34.40%) had moderate PKU and one patient (3.1 %) with mild PKU. There were positive consanguinity and family history of PKU in 87.50 % and 65.62 % of patients respectively.
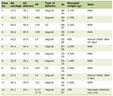 Click to view | Table 1. Demographic Data of Study Group |
As regard clinical findings, Tables 2 and 3 show that microcephaly was present in 29 cases (90.62%), fair complexion was observed in 27 cases (84.37%), and musty urine odor was observed in 23 cases (71.87%), seizures were present in 21.87%, and skin lesions in 43.75%. Neurological examination revealed hyperreflexia in 3 (9.4%) patients, tremors in 4 (12.5%) patients.
Click to view | Table 2. Clinical Data of Study Group |
Click to view | Table 3. Neuro- Radiological Complication |
Autism was present in 25% of all cases; patients were diagnosed according to DSM-IV TR criteria and classified according to the Childhood Autism Rating Scale (CARS).Table 4 and 5 show all cases diagnosed with autism (8 cases); 3 were females and 5 were males, 2 had severe autism and 6 had mild/moderate autism, 5 patients had classic PKU and 3 had moderate PKU, 3 cases had mild mental retardation and five have moderate mental retardation. All cases with autism had negative family history for autistic disorders.
Click to view | Table 4. Details for All Cases in the Study |
Click to view | Table 5. IQ Scores and Autistic Scores Among PKU Cases |
IQ scores of studied patients ranged from 34 to 72 (assessed using Stanford Binet Intelligence Scale, Fourth Edition); the mean IQ of all cases is 51.71 ± 10.94. Severemental retardation was present in 2 cases (6.25%), 14 cases (43.75%) have moderate mental retardation; 16 cases (50.00%) have mild and borderline mental retardation (Table 5). Magnetic resonance imaging (MRI) of the brain was done for all patients, periventricular increased white matter signal in T2 weighted studies was detected in 28 (87.50%) of them. The abnormalities affected mainly the occipital lobe (Table 3).
| Discussion | ▴Top |
Untreated PKU patients typically develop mental retardation, seizures, growth retardation, hyperreflexia, and skin lesions. Brain pathology of untreated patients is characterized by reduced brain weight and hypo- and demyelination of cerebral white matter. Although gray matter changes in cortical layering, tissue mass atrophy and reduced dendritic arborization have been reported, the primary neuropathologic finding of PKU is that of diffuse abnormalities within white matter. Reduction in brain size is largely due to white matter volume loss. Specific white matter abnormalities include delayed or defective myelination, diffuse white matter vacuolization (status spongiosus), demyelination, and gliosis [1, 22]. Newborn screening for PKU and the introduction of a low- Phe therapeutic diet for infants diagnosed with PKU prevents the most serious consequences [1]. In Egypt, neonatal screening is applied only for hypothyroidism and so diagnosis of PKU depends on clinical manifestations detected by experienced physicians, leading to late diagnosis of this disease in an irreversible stage.
In the present study; microcephaly, fair complexion and musty urine odor were the most frequent clinical manifestations detected in 90.62%, 84.37% and 71.87% of all cases respectively. Seizures and skin lesions were much less frequent detected in only 21.87% and 43.75% of studied patients respectively.
Baieli et al [23] studied a group of 35 patients with late diagnosis of PKU; 17 (48.5%) had microcephaly and 12 (34.2%) had seizures. Gonzalez et al [24]; reported epilepsy, tremors and hyperreflexia in 31%, 93.10 % and 27.60% respectively of their PKU series with late diagnosis. In other studies; Paine [25] observed that both skin lesions and seizures are proportionately more frequent in severely retarded patients with PKU than in those with lesser degrees of intellectual defect. In addition, the majority of these patients have blue eyes and blond hair. Mazur et al [26] observed that poor physical growth, short stature and microcephaly were the most important manifestations in their cases with untreated PKU.
In our study, the mean IQ of studied patients was 51.71, with one half of patients had moderate to severe mental retardation, and the other half had mild and borderlinemental retardation. Also the present study there were significant negative correlation between IQ and Phe level (r= -0.587, P < 0.000) Figure 1. The same result reported by Gonzalez et al [24], who also reported mild to severe mental retardation in 46.3%, borderline mental retardationin 28.5% and normal IQ scores in 25% [24]. Baieli et al [23] reported mental retardation in all patients with late diagnosis of PKU (IQ scores ranged from 37 to 49 and mean IQ 45.4).
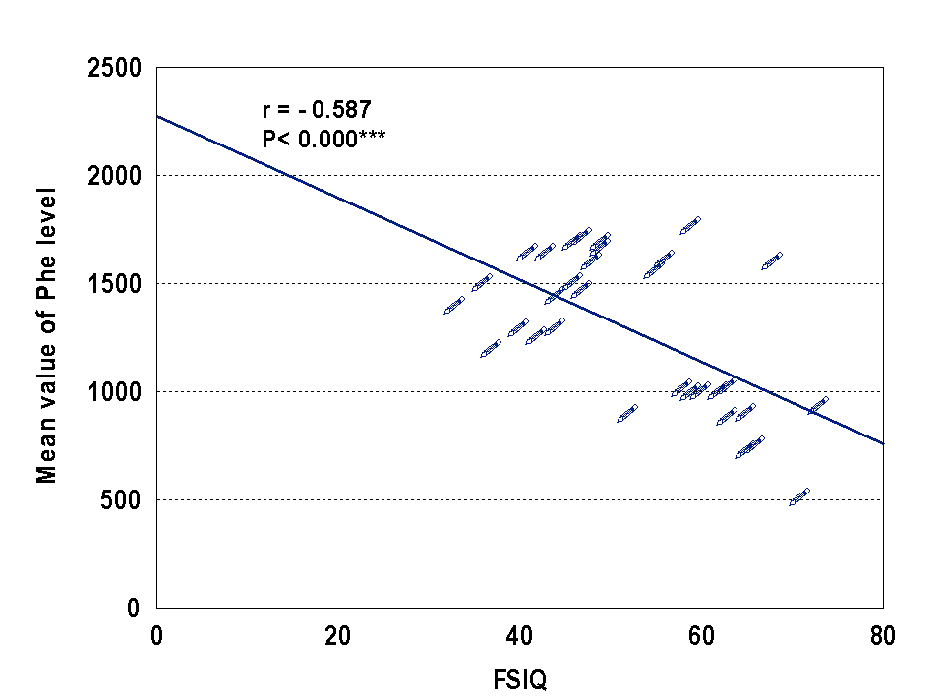 Click for large image | Figure 1. Correlation between IQ and Phe level. |
PKU was typically associated with severe intellectual impairment prior to the introduction of newborn screening and early dietary intervention. In contrast, early treated patients typically present with intellectual functioning within the normal range but often obtain lower scores compared to unaffected familial controls and unaffected non familial controls [1, 3].
The pathology underlying cognitive dysfunction in phenylketonuria is unknown, although it is clear that the high plasma concentrations of phenylalanine influence the blood-brain barrier transport of large neutral amino acids. The high plasma phenylalanine concentrations increase phenylalanine entry into brain and restrict the entry of other large neutral amino acids. Phenylalanine was found to interfere with different cerebral enzyme systems. However, apart from the neurotoxicity of phenylalanine, a deficiency of the other large neutral amino acids in brain may also be an important factor affecting cognitive function in phenylketonuria [27].
The association between PKU and autism has been documented; however, the frequency of this condition varies widely among studies [23]. Autism was present in 25% of all cases in our study that of all cases diagnosed with autism (8 cases); 3 were females and 5 were males, 2 had severe autism and 6 had mild/moderate autism, 5 patients had classic PKU and 3 had moderate PKU. In agreement with our results; Knobloch and Pasamanick [28]reported autism in 14 cases (21.8%) of children with PKU. Hackney et al [29] identified 9 patients (19.56%) with autism out of 46 PKU patients. Friedman [30] described over 40 cases with PKU and autism in a review article. Lowe et al [11] identified 2 autistic patients with PKU. More recent studies including Baieli et al [23], who reported that 5.7% of late diagnosed PKU cases had autism. Halayem-Dhouib [31] reported 14 children were suffering from autism in a series of 19 PKU patients.
The link between poor control/ late diagnosis PKU and autism suggests that with improvements in diagnosis and treatment of PKU, the association with autism will likely decrease. The association between autism in PKU has important implications for understanding possible underlying factors in the development of autism. In the case of PKU, the toxic levels of phenylalanine hydroxylase appear to play a significant role in the development of autism symptomatology [14].
Magnetic resonance imaging (MRI) of the brain was done for all patients, periventricular increased white matter signal in T2 weighted studies was detected in 28 (87.50%) of them. Previous studies reported MRI abnormalities in 70 to 100 % of cases with PKU [24, 32, 33]. White matter abnormalities tend to be associated with age and metabolic control such that the prevalence of these abnormalities is higher and more severe in older children, those who are off treatment or those with high Phe levels. Researches had also demonstrated that in severe cases with PKU, the white matter abnormalities can extend into subcortical regions, internal capsule, brain stem and cerebellum. The white matter pathology observed in PKU is generally diffuse and, as a consequence, multiple pathways may be compromised and mild to moderate deficits may be observed over a range of functions including motor skills and coordination, visual functioning, processing speed, language, memory and learning, as well as attention and executive functioning. Consistent with this posit, a broad array of neuropsychological difficulties had been reported in the PKU population [32-34].
Conclusion
Delay in the diagnosis and management of PKU leads to very high incidence of intellectual disabilities and autistic features in these patients.
| Recommendations | ▴Top |
1), neonatal screening for PKU is highly recommended for early detection and management; 2), further studies are needed to answer the question of whether PKU patients have a specific autistic phenotype or whether their behavior represents the evolution of autism with age with late diagnosis.
| References | ▴Top |
- Scriver CR, Kaufman S. Hyperphenylalaninemia: phenylalanine hydroxylase deficiency. In: Scriver CR, Beaudet AL, et al, editors. The Metabolic and Molecular Bases of Inherited Disease. 8th ed. New York: McGraw-Hill; 2001; p. 1667-1724.
- Campistol J, Gassio R, Artuch R, Vilaseca MA. Neurocognitive function in mild hyperphenylalaninemia. Dev Med Child Neurol. 2011;53(5):405-408.
doi pubmed - Blau N, van Spronsen FJ, Levy HL. Phenylketonuria. Lancet. 2010;376(9750):1417-1427.
doi - Brumm VL, Bilder D, Waisbren SE. Psychiatric symptoms and disorders in phenylketonuria. Mol Genet Metab. 2010;99(Suppl 1):S59-63.
doi pubmed - Williams RA, Mamotte CD, Burnett JR. Phenylketonuria: an inborn error of phenylalanine metabolism. Clin Biochem Rev. 2008;29(1):31-41.
pubmed - Brumm VL, Azen C, Moats RA, Stern AM, Broomand C, Nelson MD, Koch R. Neuropsychological outcome of subjects participating in the PKU adult collaborative study: a preliminary review. J Inherit Metab Dis. 2004;27(5):549-566.
doi pubmed - Surtees R, Blau N. The neurochemistry of phenylketonuria. Eur J Pediatr. 2000;159(Suppl 2):S109-113.
doi pubmed - Scarabino T, Popolizio T, Tosetti M, Montanaro D, Giannatempo GM, Terlizzi R, Pollice S, et al. Phenylketonuria: white-matter changes assessed by 3.0-T magnetic resonance (MR) imaging, MR spectroscopy and MR diffusion. Radiol Med. 2009;114(3):461-474.
doi pubmed - Kuehn BM. CDC: autism spectrum disorders common. JAMA. 2007;297(9):940.
doi pubmed - Manzi B, Loizzo AL, Giana G, Curatolo P. Autism and metabolic diseases. J Child Neurol. 2008;23(3):307-314.
doi pubmed - Lowe TL, Tanaka K, Seashore MR, Young JG, Cohen DJ. Detection of phenylketonuria in autistic and psychotic children. JAMA. 1980;243(2):126-128.
doi pubmed - Chen CH, Hsiao KJ. A Chinese classic phenylketonuria manifested as autism. Br J Psychiatry. 1989;155:251-253.
doi - Steiner CE, Acosta AX, Guerreiro MM, Marques-de-Faria AP. Genotype and natural history in unrelated individuals with phenylketonuria and autistic behavior. Arq Neuropsiquiatr. 2007;65(2A):202-205.
doi pubmed - Moss J, Howlin P. Autism spectrum disorders in genetic syndromes: implications for diagnosis, intervention and understanding the wider autism spectrum disorder population. J Intellect Disabil Res. 2009;53(10):852-873.
doi pubmed - Caglayan AO. Genetic causes of syndromic and non-syndromic autism. Dev Med Child Neurol. 2010;52(2):130-138.
doi pubmed - Gillberg C, Coleman M. The biology of the autistic syndromes. 3rd ed. 2000,.
- Melika LK. The Stanford Binet Intelligence Scale: fourth Edition: Arabic Examiner's Handbook. Dar El-Maref Publishing, Egypt, Cairo, 1998.
pubmed - Roeleveld N, Zielhuis GA, Gabreels F. The prevalence of mental retardation: a critical review of recent literature. Dev Med Child Neurol. 1997;39(2):125-132.
doi pubmed - American Psychiatric Association. Diagnostic and statistical manual of mental disorders 4th ed., DSM-IV. Washington, DC: 1994.
- Schopler E., Reichller R. & Renner BR. The Childhood Autism Rating Scale CARS, 1994; Western Psychological Services, 12031 Wilshire Boulevard, Los Angeles, California, 90025-91251.
- Rellini E, Tortolani D, Trillo S, Carbone S, Montecchi F. Childhood Autism Rating Scale (CARS) and Autism Behavior Checklist (ABC) correspondence and conflicts with DSM-IV criteria in diagnosis of autism. J Autism Dev Disord. 2004;34(6):703-708.
doi pubmed - Hahnel S. Brain MRI Abnormalities in Phenylketonuria - An Update on MR Imaging and MR Volumetry. Clin Neuroradiol. 2008; 18:19-24.
doi - Baieli S, Pavone L, Meli C, Fiumara A, Coleman M. Autism and phenylketonuria. J Autism Dev Disord. 2003;33(2):201-204.
doi pubmed - Gonzalez MJ, Gutierrez AP, Gassio R, Fuste ME, Vilaseca MA, Campistol J. Neurological complications and behavioral problems in patients with phenylketonuria in a follow-up unit. Mol Genet Metab. 2011;104(Suppl):S73-79.
doi pubmed - Paine RS. The variability in manifestations of untreated patients with phenylketonuria (phenylpyruvic aciduria). Pediatrics. 1957;20(2):290-302.
pubmed - Mazur A, Jarochowicz S, Sykut-Cegielska J, Gradowska W, Kwolek A, Oltarzewski M. Evaluation of somatic development in adult patients with previously undiagnosed and/or untreated phenylketonuria. Med Princ Pract. 2010;19(1):46-50.
doi pubmed - van Spronsen FJ, Hoeksma M, Reijngoud DJ. Brain dysfunction in phenylketonuria: is phenylalanine toxicity the only possible cause? J Inherit Metab Dis. 2009;32(1):46-51.
doi pubmed - Knobloch H, Pasamanick B. Some etiologic and prognostic factors in early infantile autism and psychosis. Pediatrics. 1975;55(2):182-191.
pubmed - Hackney IM, Hanley WB, Davidson W, Lindsao L. Phenylketonuria: mental development, behavior, and termination of low phenylalanine diet. J Pediatr. 1968;72(5):646-655.
doi - Friedman E. The "autistic syndrome" and phenylketonuria. Schizophrenia. 1969;1:249-261.
- Halayem-Dhouib S, Khmir S, Belhaj A et al. Autistic symptoms in children with phenylketonuria. Neuropsychiatr Enfance Adolesc. 2012; 60(5)S209.
- Moller HE, Weglage J, Bick U, Wiedermann D, Feldmann R, Ullrich K. Brain imaging and proton magnetic resonance spectroscopy in patients with phenylketonuria. Pediatrics. 2003;112(6 Pt 2):1580-1583.
pubmed - Leuzzi V, Tosetti M, Montanaro D, Carducci C, Artiola C, Antonozzi I, Burroni M, et al. The pathogenesis of the white matter abnormalities in phenylketonuria. A multimodal 3.0 tesla MRI and magnetic resonance spectroscopy (1H MRS) study. J Inherit Metab Dis. 2007;30(2):209-216.
doi pubmed - Thompson AJ, Tillotson S, Smith I, Kendall B, Moore SG, Brenton DP. Brain MRI changes in phenylketonuria. Associations with dietary status. Brain. 1993;116(Pt 4):811-821.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Neurology Research is published by Elmer Press Inc.