

| Journal of Neurology Research, ISSN 1923-2845 print, 1923-2853 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Neurol Res and Elmer Press Inc |
| Journal website http://www.neurores.org |
Case Report
Volume 2, Number 6, December 2012, pages 250-254
Childhood Ataxia With Central Nervous System Hypomyelination: A Report of First Two Cases With Juvenile and Adult - Onset Forms in a Turkish Family
Gulay Kenangila, c, Destina Yalcinb
aNeurology Department, Erenkoy Education and Research Hospital for Psychiatric and Neurological Disorders, Istanbul, Turkey
bNeurology Department, Umraniye Education and Research Hospital, Istanbul, Turkey
cCorresponding author: Gulay Kenangil, Yenisehir mah. Kagnici yolu sok. No: 50 5, 7000-A 320 Atasehir, Istanbul, Turkey
Manuscript accepted for publication December 19, 2012
Short title: Childhood Ataxia
doi: https://doi.org/10.4021/jnr162w
| Abstract | ▴Top |
Childhood ataxia with central nervous system hypomyelination (CACH)) is an inherited autosomal recessive white matter disease which is also known as leukoencephalopathy with vanishing white matter (VWM). Ataxia and spasticity are the predominant symptoms of the disease. It is a progressive disease with exacerbations. Manyetic resonance imaging (MRI) is diagnostic with diffuse white matter abnormalities. It can be seen at all ages but adult forms are rare. We report here the first two cases of juvenile and adult-onset forms of the disease in Turkey. They are two sisters with age of 21 and 25 who presented us with gait difficulty and spasticity. They had seizures and also the typical MRI findings of CACH. We should consider it in the differential diagnosis of gait difficulty, ataxia and spasticity in adult patients.
Keywords: Ataxia; White matter disease; Central nervous system; Hypomyelination
| Introduction | ▴Top |
Childhood ataxia with central nervous system hypomyelination (CACH)) is an inherited autosomal recessive white matter disease which is also known as leukoencephalopathy with vanishing white matter (VWM). The first pathological description of the disease was made in 1962 by Eicke in a 36- year-old woman with gait difficulties and secondary amenorrhea [1] and it was rediscovered by Hanefeld and colleagues in 1993 and Schiffman in 1994 as a clinical entity characterized by childhood-onset progressive leukoencephalopathy with an autosomal recessive mode of inheritance [2, 3]. Diagnostic criteria are normal early motor and mental development with a chronic progressive or episodic course of neurological deterioration following infection or trauma, presence of cerebellar ataxia and spasticity as evident clinical symptoms and the Magnetic resonance imaging (MRI) findings of bilateral cerebral white matter involvement and varying degrees of cerebellar atrophy [4-7].
There are identifiable mutations in five causative genes (EIF2B1, EIF2B2, EIF2B3, EIF2B4, EIF2B5) encoding the five subunits of eucaryotic translation initiation factor 2B (EIF2B) in 90% of individuals [8]. A broad clinical spectrum from congenital to adult-onset forms has been described [5, 9]. We present here two sisters of age 21 and 25 years, presenting with juvenile and adult - onset forms of the disease.
| Case Report | ▴Top |
Case 1
This 25-year-old woman was the second child of a nonconsanguinoeus marriage, with normal motor and mental development. Her symptoms began with progressive fatigue and ataxia at age 20. She didn’t have any paresthetic complains, sensory loss, radicular pain or sphincter abnormalities. She had received education for 8 years and fully independent in all daily activities. She did not have any history of head trauma, febrile illness, cardiac symptoms or seizure. Her older brother was asymptomatic while her younger sister was strikingly affected. Her parents did not have any illness other than the diabetes mellitus of her father.
On general examination, she had scoliosis and pes planus. On neurological examination, she had 30/30 on mini-mental state examination (MMSE). Fundus examination did not reveal any abnormality. She did not have any nystagmus or gaze abnormalities. She had mild paresis in both proximal lower extremities, so she had difficulty raising from the ground. She had mild spasticity which was more prominent while walking. Cerebellar tests were normal. Her deep tendon reflexes were brisk. Right plantar response was extensor. She had mild ataxia while walking and was getting worse with fatigue.
Routine hematological and biochemical investigations, electrocardiogram and echocardiography were normal. Nerve conduction studies failed to reveal any abnormalities.
MRI demonstrated diffuse symmetrical white matter hypointensities on T1-weighted and hyperintensities on T2- weighted sequences with arcuate fibre involvement. Lateral ventricules were dilated due to periventricular white matter loss (Fig. 1).
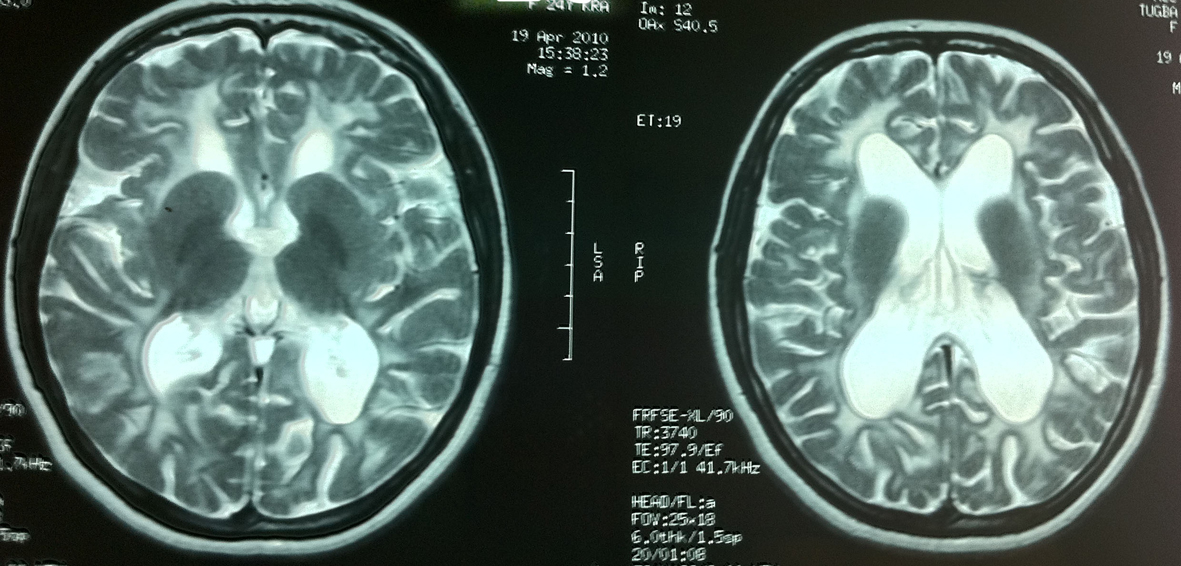 Click for large image | Figure 1. Diffuse white matter involvement on T2-weighted images of patient 1. |
On EEG examination the background activity was composed of 7 - 7.5 Hz theta waves. During hyperventilation, generalized 2.5 Hz spike and wave discharges lasting 2 - 4 seconds were recorded several times. Some of these episodes were clinically accompanied by interruption of number counting and staring. The patient was not aware of these absences and denied any antiepileptic treatment.
Gynecologic ultrasonography (USG) was normal.
Case 2
This 21-year-old girl with a first generalized seizure at age 13, had gait difficulty and ataxia since that time. She had received education for 8 years. Her school performance was above average. She had totally three generalized seizures before treatment and used valproic acid for 3 years. Her drug therapy was stopped five years ago. She has remained seizure free without any recurrence of epileptic seizures till now.
She had also pes planus and mild scoliosis like her sister. She got 30/30 on MMSE. Fundus examination did not reveal any abnormality. She did not have any nystagmus or gaze abnormalities. She had mild paresis in both proximal lower extremities, so she had difficulty raising from the ground. She had mild spasticity which is more prominent while walking. Deep tendon reflexes were increased. Her plantar reflexes were bilaterally extensor. She had dysdiadochokinesia on the left side and intention tremor on the right side. Her gait was ataxic and spastic.
Her MRI demonstrated diffuse white matter abnormalities almost similar to those of her sister (Fig. 2). On EEG she had diffuse theta activity. Gynecologic USG was normal.
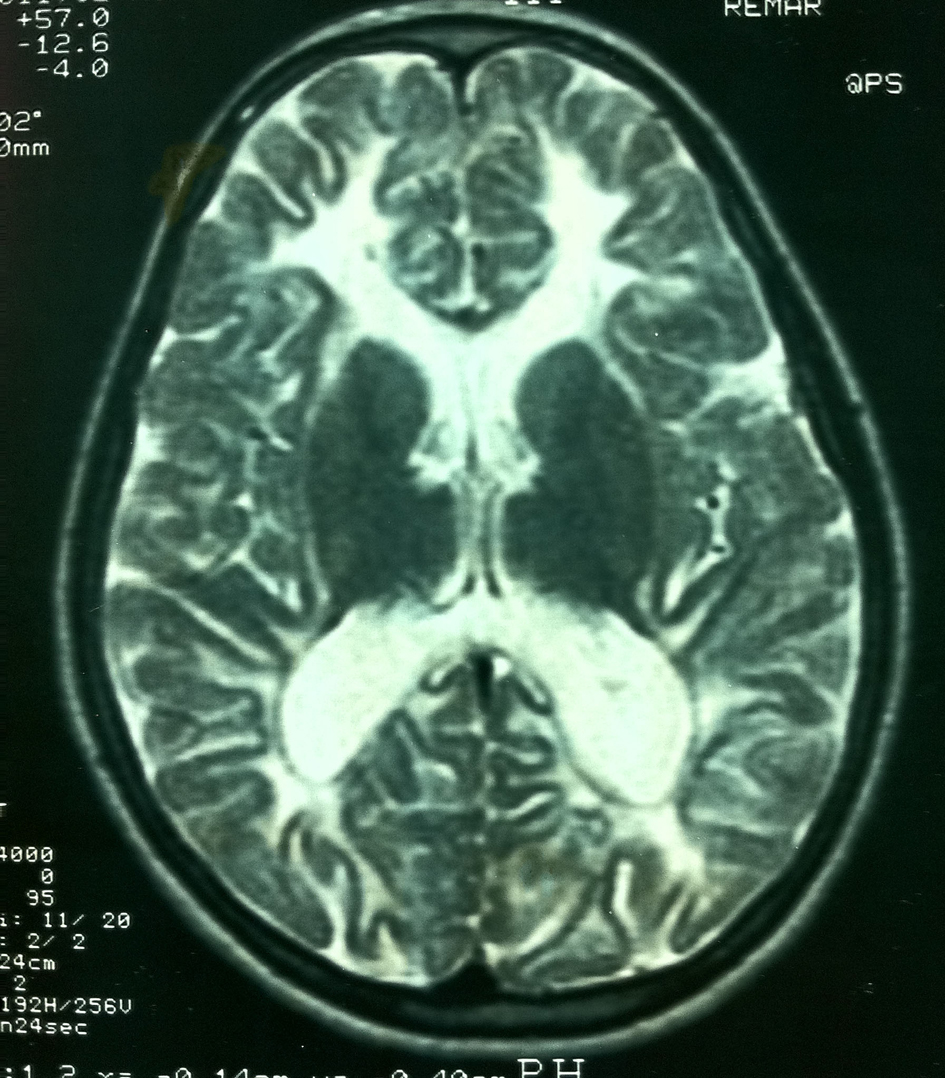 Click for large image | Figure 2. Diffuse white matter involvement on T2-weighted images of patient 2 similar to that of her sister. |
| Discussion | ▴Top |
In this report, we have presented two siblings of a Turkish family, affected by CACH with characteristic MRI findings and clinical features. These cases are the first reported juvenile and adult forms of the disease in Turkey.
In CACH there are different phenotypes ranging from a prenatal/congenital form to a subacute infantile form (onset of age < 1 year), an early childhood-onset form (onset of age 1 - 5 years), a late childhood/juvenile-onset form (onset of age 5 - 15 years), and an adult-onset form [5]. Early infantile form is characterized by severe encephalopathy, oligohydramnios, intrauterine growth retardation, microcephaly, contractures, cataract, pancreatitis, hepatosplenomegaly, and renal hypoplasia. Progressive megalencephaly also has been reported in three children with an infantile form of CACH who had protracted disease course that deteriorated after viral infections [10]; Our patients neither had microcephaly nor were infants or have had any episode of deterioration till now.
Encephalopathy is more severe in the early-onset forms and seizures are predominant clinical feature [8]. In all later-onset forms, initial motor and mental development is normal. Both our patients had normal mental and motor development. Our patient with a juvenile-onset form had seizures which stopped 5 years ago without any recurrence. The adult-onset case also had absence seizures with EEG abnormalities.
In juvenile-onset form, children develop symptoms between 5 and 15 years of age. They have a more slowly progressive spastic diplegia, relative sparing of cognitive ability and likely long-term survival with long periods of stability [3, 5]. The symptoms of our juvenile-onset patient began at age 13, which were slowly progressive without any cognitive deficit.
Cognitive decline or behavioral problems may be the presenting symptom in adult- onset forms of the disease [11, 12]. Since 1998, milder variants of the disease with an adolescent or adult onset have been identified [5]. The symptoms began at age 20 in our adult-onset patient who had no cognitive or behavioral problems and may be an example for the milder form of the adult-onset disease.
Neurological deterioration has a chronic progressive or subacute course. Episode of subacute deterioration may follow minor febrile infection, minor head trauma or fright which may lead to lethargy or coma [4, 5, 13]. Our patients did not experience any traumas or stress before the appearance of clinical symptoms. The episodes of rapid deterioration are typically less substantial in patients with a later onset [1, 14]. Both of our patients had mild spasticity, pyramidal signs, mild cerebellar signs with normal motor and mental development until late childhood and adulthood, confirming the typical course of the disease in juvenile and adult- onset forms. The progression in our patients is slow and without any exacerbations as yet.
Ovaries are the next most commonly involved organs and ovarian dysgenesis may cause primary or secondary amenorrhea [4, 15, 16]; Both of our patients did not have any ovarian pathology.
Besides all these, it is intriguing that both our patients had pes planus and scoliosis which are not noted as classical signs in CACH. They also had seizures, although they are rare in juvenile and adult-onset forms.
Van der Knaap reported that the diagnosis of CACH can be made with confidence in individuals with typical clinical findings and characteristic abnormalities on cranial MRI [14].
MRI of the brain is usually diagnostic in CACH. It shows an abnormal signal of all or almost all cerebral white matter [4, 5, 17]. In the beginning, the U-fibres may be relatively spared; but as sequential MRIs reveal increase in CSF-like white matter areas, signal abnormalities of U- fibres and some cortical atrophy, Van der Knaap termed this as vanishing white matter [4]. MRI shows evidence of progressive rarefaction and cystic degeneration of the affected white matter, which is replaced by fluid [4, 5]. This change is best shown by proton density and fluid-attenuated inversion recovery (FLAIR) images. In the series of Labauge, cystic changes included the corpus callosum [11]. Contrast enhancement has never been reported.
In our patients, MRI showed diffuse symmetrical white matter abnormalities involving the arcuate fibres. We also observed cortico-subcortical atrophy with ventricular dilatation.
Several presymptomatic and mildly symptomatic individuals underwent MRI, which always revealed extensive cerebral white-matter abnormalities, although not necessarily with evidence of rarefaction or cystic degeneration at first [4, 18]. We could not perform MRI scans for the other asymptomatic members of the family.
The true prevalence of CACH is not known; it is considered one of the most common leukodystrophies. It was the most common type in a study of unclassified leukodystrophies of childhood [19].
Acute demyelinating encephalomyelitis and encephalitis are the first to be considered in the differential diagnosis of patients presenting with rapid neurological deterioration after a febrile infection. MRI usually shows multifocal white matter lesions with an asymmetrical distribution in acute demyelinating encephalomyelitis and variable lesions involving both white and grey matter in encephalitis [14]. Contrast enhancement and restricted diffusion in affected areas are features of both acute demyelinating encephalomyelitis and encephalitis, but not of CACH. It can be differentiated from megalencephalic leukoencephalopathy with subcortical cysts (MLSC) by the age of onset and the temporo-frontal cysts observed in MLSC [6]. X-linked adrenoleukodystrophy, metachromatic leukodystrophy, Krabbe and Canavan disease are the other white matter diseases which also may be considered in differential diagnosis of CACH [17]. The characteristic MRI findings with cystic degeneration help to distinguish CACH from these disorders because as a rule none have cystic degeneration. In addition, Canavan disease has elevated N- acetyl-aspartate (NAA) in magnetic resonance spectroscopy (MRS) [6]. We can also think of Alexander disease but it has a frontal predominance and prominent basal ganglia involvement with contrast enhancement. Mitochondrial disorders such as pyruvate dehydrogenase and pyruvate carboxylase deficiencies may mimic the MRI features of CACH, whereas clinically there is motor and mental delay in these disorders, contrary to CACH [20, 21].
Lactate and pyruvate levels may help in differential diagnosis. Because of the typical MRI and clinical findings we did not examine the lactate and pyruvate levels. In addition, the decrease in the ratio of cerebrospinal fluid (CSF) asialo-transferrine to total transferrine may be a biomarker for distinguishing the patients with EIF2B mutation. It is a fast (< 48 hours) and a cheap technic before the genetic evaluation. We do not have this technic in our country.
There is no specific treatment for CACH [14]. Stress situations which provoke deterioration in VWM patients must be avoided. Although not sufficient to prevent the onset or progression of the disease, use of antibiotics and antipyretics, vaccinations, and abstinence of contact sports are simple but important measures.
As a consequence of recent research findings on genetics of the disease, prenatal diagnosis for disease-causing mutations has become available for families. Two-thirds of the patients with CACH have mutations in EIF2B5, which is the largest subunit. It was also shown that if the MRI was typical for CACH, mutations were almost invariably found [14.] We could not perform a genetic analysis for our patients but the typical clinical and MRI findings helped us for the diagnosis.
In conclusion, CACH is not an uncommon white matter disease and we should consider it in the differential diagnosis of gait difficulty, ataxia and spasticity in adult patients. We also should be aware of its classical MRI findings in order to recognize it.
| References | ▴Top |
- Eicke WJ. Polycystische umwandlung des marklagers mitprogredientem verlauf. Atypische diff use sklerose? Arch Psychiat Nervenkr 1962; 203: 599-602.
doi - Hanefeld F, Holzbach U, Kruse B, Wilichowski E, Christen HJ, Frahm J. Diffuse white matter disease in three children: an encephalopathy with unique features on magnetic resonance imaging and proton magnetic resonance spectroscopy. Neuropediatrics. 1993;24(5):244-248.
doi pubmed - Schiffmann R, Moller JR, Trapp BD, Shih HH, Farrer RG, Katz DA, Alger JR, et al. Childhood ataxia with diffuse central nervous system hypomyelination. Ann Neurol. 1994;35(3):331-340.
doi pubmed - van der Knaap MS, Barth PG, Gabreels FJ, Franzoni E, Begeer JH, Stroink H, Rotteveel JJ, et al. A new leukoencephalopathy with vanishing white matter. Neurology. 1997;48(4):845-855.
doi pubmed - van der Knaap MS, Kamphorst W, Barth PG, Kraaijeveld CL, Gut E, Valk J. Phenotypic variation in leukoencephalopathy with vanishing white matter. Neurology. 1998;51(2):540-547.
doi pubmed - Ravishankar S, Sinha S, Taly AB, Panicker J. Vanishing white matter disease: Phenotypic, MR imaging and H spectroscopic obsevations. Ann Indian Acad Neurol 2006;9:172-4.
doi - Vaidya SR, Desai SB, Khadilkar SV, Mehta NA. Childhood ataxia with cerebral hypomyelination (CACH) syndrome: a study of three siblings. Neurol India. 2004;52(3):372-374.
pubmed - van der Knaap MS, Leegwater PA, Konst AA, Visser A, Naidu S, Oudejans CB, Schutgens RB, et al. Mutations in each of the five subunits of translation initiation factor eIF2B can cause leukoencephalopathy with vanishing white matter. Ann Neurol. 2002;51(2):264-270.
doi pubmed - Fogli A, Boespflug-Tanguy O. The large spectrum of eIF2B-related diseases. Biochem Soc Trans. 2006;34(Pt 1):22-29.
pubmed - Passemard S, Gelot A, Fogli A, N'Guyen S, Barnerias C, Niel F, Doummar D, et al. Progressive megalencephaly due to specific EIF2Bepsilon mutations in two unrelated families. Neurology. 2007;69(4):400-402.
doi pubmed - Labauge P, Horzinski L, Ayrignac X, Blanc P, Vukusic S, Rodriguez D, Mauguiere F, et al. Natural history of adult-onset eIF2B-related disorders: a multi-centric survey of 16 cases. Brain. 2009;132(Pt 8):2161-2169.
doi pubmed - Denier C, Orgibet A, Roffi F, Jouvent E, Buhl C, Niel F, Boespflug-Tanguy O, et al. Adult-onset vanishing white matter leukoencephalopathy presenting as psychosis. Neurology. 2007;68(18):1538-1539.
doi pubmed - Vermeulen G, Seidl R, Mercimek-Mahmutoglu S, Rotteveel JJ, Scheper GC, van der Knaap MS. Fright is a provoking factor in vanishing white matter disease. Ann Neurol. 2005;57(4):560-563.
doi pubmed - van der Knaap MS, Leegwater PA, van Berkel CG, Brenner C, Storey E, Di Rocco M, Salvi F, et al. Arg113His mutation in eIF2Bepsilon as cause of leukoencephalopathy in adults. Neurology. 2004;62(9):1598-1600.
doi pubmed - Fogli A, Rodriguez D, Eymard-Pierre E, Bouhour F, Labauge P, Meaney BF, Zeesman S, et al. Ovarian failure related to eukaryotic initiation factor 2B mutations. Am J Hum Genet. 2003;72(6):1544-1550.
doi pubmed - Schiffmann R, Tedeschi G, Kinkel RP, Trapp BD, Frank JA, Kaneski CR, Brady RO, et al. Leukodystrophy in patients with ovarian dysgenesis. Ann Neurol. 1997;41(5):654-661.
doi pubmed - Schiffmann R, van der Knaap MS. Invited article: an MRI-based approach to the diagnosis of white matter disorders. Neurology. 2009;72(8):750-759.
doi pubmed - van der Knaap MS, Pronk JC, Scheper GC. Vanishing white matter disease. Lancet Neurol. 2006;5(5):413-423.
doi - van der Knaap MS, Breiter SN, Naidu S, Hart AA, Valk J. Defining and categorizing leukoencephalopathies of unknown origin: MR imaging approach. Radiology. 1999;213(1):121-133.
pubmed - Senol U, Haspolat S, Karaali K, Luleci E. MR imaging of vanishing white matter. AJR Am J Roentgenol. 2000;175(3):826-828.
pubmed - de Lonlay-Debeney P, von Kleist-Retzow JC, Hertz-Pannier L, Peudenier S, Cormier-Daire V, Berquin P, Chretien D, et al. Cerebral white matter disease in children may be caused by mitochondrial respiratory chain deficiency. J Pediatr. 2000;136(2):209-214.
doi
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Neurology Research is published by Elmer Press Inc.