

| Journal of Neurology Research, ISSN 1923-2845 print, 1923-2853 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Neurol Res and Elmer Press Inc |
| Journal website http://www.neurores.org |
Case Report
Volume 2, Number 3, June 2012, pages 115-118
Partial Deletion of EMD Gene in a Patient With Emery-Dreifuss Muscular Dystrophy
Adela Escuderoa, Paloma Martinez-Monteroa, Luis Fernandeza, Pilar Carrascoa, Luis Garcia-Gueretab, Samuel I.Pascual-Pascualc, Manuel Gutierrez-Molinad, Jesus Molanoa, e
aINGEMM/IdiPAZ/Hospital Universitario La Paz, Spain
bCardiology Department, Hospital Universitario La Paz, Spain
cNeuropediatrics Department/Hospital Universitario La Paz, Spain
dPathology Department/Hospital Universitario La Paz, Spain
eCorresponding author: Jesus Molano, Unidad de Genetica Molecular. INGEMM/IdiPAZ/Hospital Universitario La Paz. Paseo de la Castellana 261; 28046 Madrid. Spain
Manuscript accepted for publication June 19, 2012
Short title: Partial Deletion of EMD Gene
doi: https://doi.org/10.4021/jnr107w
| Abstract | ▴Top |
Emery-Dreifuss muscular dystrophy (EDMD) is a genetic disease that can be inherited as autosomal dominant, autosomal recessive or X-linked form. The recessive X-linked form of EDMD is caused by mutations in the EMD gene. Immunohistochemistry of muscle biopsy of the patient with specific antibodies fordystrophin, sarcoglycans and emerin. DNA analysis of the patient and his relatives include homemade Multiplex Ligation-dependent Probe Amplification (MLPA) analysis, Extended Polymerase Chain Reaction (Long PCR) and DNA sequencing. We present a case of a 16-year-old male patient with pacemaker implantation due to atrioventricular block. Muscle biopsy showed immunohistochemistry pattern negative for emerin. DNA analysis showed a deletion of 2640 bp expanding from the 5’ region, including the 5’-UTR and exon 1 and part of exon 2 of EMD gene. The new mutation is the fifth large deletion reported in the EMD gene and is associated to early cardiac symptoms.
Keywords: EMD gene; Emery-Dreifuss muscular dystrophy; Muscular dystrophies; Atrioventricular block
| Introduction | ▴Top |
Emery-Dreifuss muscular dystrophy (EDMD) is a genetic disease that can be inherited as autosomal dominant, autosomal recessive or X-linked form. The recessive X-linked form of EDMD is caused by mutations in the EMD that encodes the emerin protein [1, 2] and the dominant X-linked form is due to mutations inFHL1 gene that encodes four-and-a-half-LIM 1 protein [3, 4]. The most common cause of autosomal dominant EDMD are mutations in the LMNA/C gene [5, 6]although in some cases autosomal dominant EDMD may also be caused by mutations in the SYNE2 gene [7]. Recently, Liang et al, [8] have reported two cases of autosomal dominant EDMD caused by mutations in the TMEM43 gene. Some cases of autosomal recessive EDMD have been reported due to mutations in theSYNE1 gene [7].
| Case Report | ▴Top |
We present a case of a 16-year-old male patient (ED25) with intravenous pacemaker sequential implantation due to atrioventricular block of advanced degree. Informed consent was obtained from the patient and relatives. Genomic DNA was extracted from peripheral blood leukocytes by standardized procedures (commercial Gentra Puregene Blood Kit from Qiagen, Maryland, USA). Intronic primers were designed to amplify exons and flanking intronic sequences of EMD and LMNA/C genes. Amplified DNA products were sequenced by the di-deoxy termination method (BigDye Sequencing Kit version 3.1 from Applied Biosystems, CA, USA) using an ABI 3130 DNA sequencer. Both strands of DNA were sequenced. Specific Multiplex Ligation-dependent Probe Amplification (MLPA) probes were designed to detect deletions and duplications in the EMD gene [9] (specific probe sequences are available from corresponding author upon request). Long PCR was carried out using Expand 20KbPLUS PCR System (Roche Diagnostics GmbH Mannheim, Germany) and oligonuclotide primers, 5’EMD-FOR (GAGGGCTTCGTGGGGGAGTC) and GEMD3R2 (5’-CTGTGAGCGGGGTCCCTGAC), following manufacturer instructions.
Patient ED25 presented moderate muscular weakness since he was 3 year-old, with no disease progress. From the age of 10 he developed mild contractures of elbows, hips and knees, and atrioventricular block. Serum CK determinations during the evolution ranged from 600 to 1300 U (normal maximum 150). The first muscle biopsy, performed at 7 years of age showed nonspecific myopathic disorders (emerin protein could not be analyzed). A new muscle biopsy at 16 years of age showed mild myopathic changes with variability of the fibers without altering the general muscle architecture with minimal fatty infiltration. Immunohistochemistry patterns with specific antibodies were irregular for dystrophin and sarcoglycans and negative for emerin. At this age, physical examination showed mild facial weakness, joint contractures in elbows and hips, mild shortening of Achilles tendon and mild suprascapular atrophy. The patient was able to run and jump. Mean and minimal heart rate from 24-h Holter recordings was 52 and 32 heartbeats per minute, respectively. The provisional diagnosis was Emery-Dreifuss muscular dystrophy.
The patient was analyzed for routine blood testing. Blood CK was repeatable high (around 1300 IU/L). Arterial blood oxygen saturation was low (63% saturation) andCO2 concentration was increased (30.8 mM; normal values from 23 to 27 mM). Electromyogram showed minimal changes.
Patient’s DNA was sequenced for exons and exon/intron regions of the EMD and LMNA/C genes. After several trials to PCR amplify exons 1 and 2 of EMD gene without success we postulated the existence of a deletion affecting the 5’ region of the gene. To test this hypothesis, and to establish the condition of carrier/non-carrier of the patient’s mother and sister, we designed a Multiplex Ligation-dependent Probe Amplification assay (MLPA) specific for EMD gene. As shown inFigure 1A, the propositus presents a deletion of exon 1 and 2 of EMD gene. His mother and sister are carriers for Emery-Dreifuss muscular dystrophy since they carry the same deletion in one allele (Fig. 1B). To establish the extension of the deletion in the patient we amplified, using different sets of primers, a fragment of 2206 bp while the expected size for normal allele is 4846 bp. The selected primers were 5’EMD-FOR and GEMDR2. We sequenced the deleted allele to determine the breakpoint of the deletion. Sequence of the breakpoint region is shown in Figure 2A. Deletion expands 2640 nucleotides, from exon 2 to 2372 nucleotides upstream of the initial ATG of EMD gene (c.1-2375_EMD:145del) (Fig. 2B).
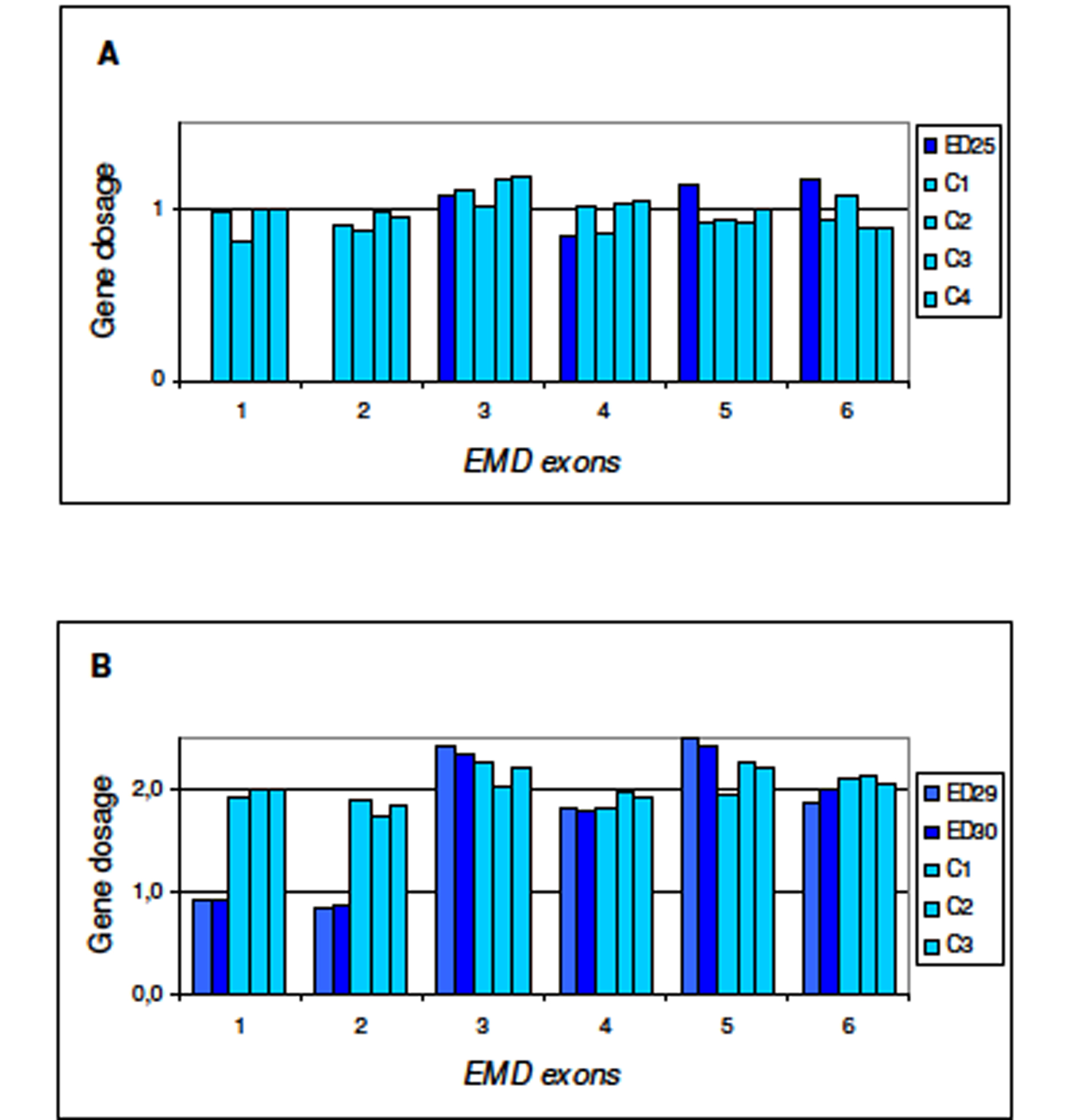 Click for large image | Figure 1. Multiplex Ligation-dependent Probe Amplification (MLPA) for dosage of EMD gene. Procedure for test performance was the one described by Shouten et al. [http://www.neurores.org/index.php/neurores/article/view/107/125 9]. For data analysis a global normalisation was achieved by dividing the peak area for each amplification product by the average peak area of all peaks in that sample. Normalised peak areas were compared to the results obtained for several control samples. In the final step, the results obtained were visualised using Excel graphics. In abscissa exons 1 to 6 of EMD gene and in ordinate gene dosage as deduced from peak areas. A) ED25 is the propositus; C1, C2, C3, and C4 are male controls. B) ED29 and ED30 are the mother and sister of the propositus, respectively; C1, C2, C3 are female controls. |
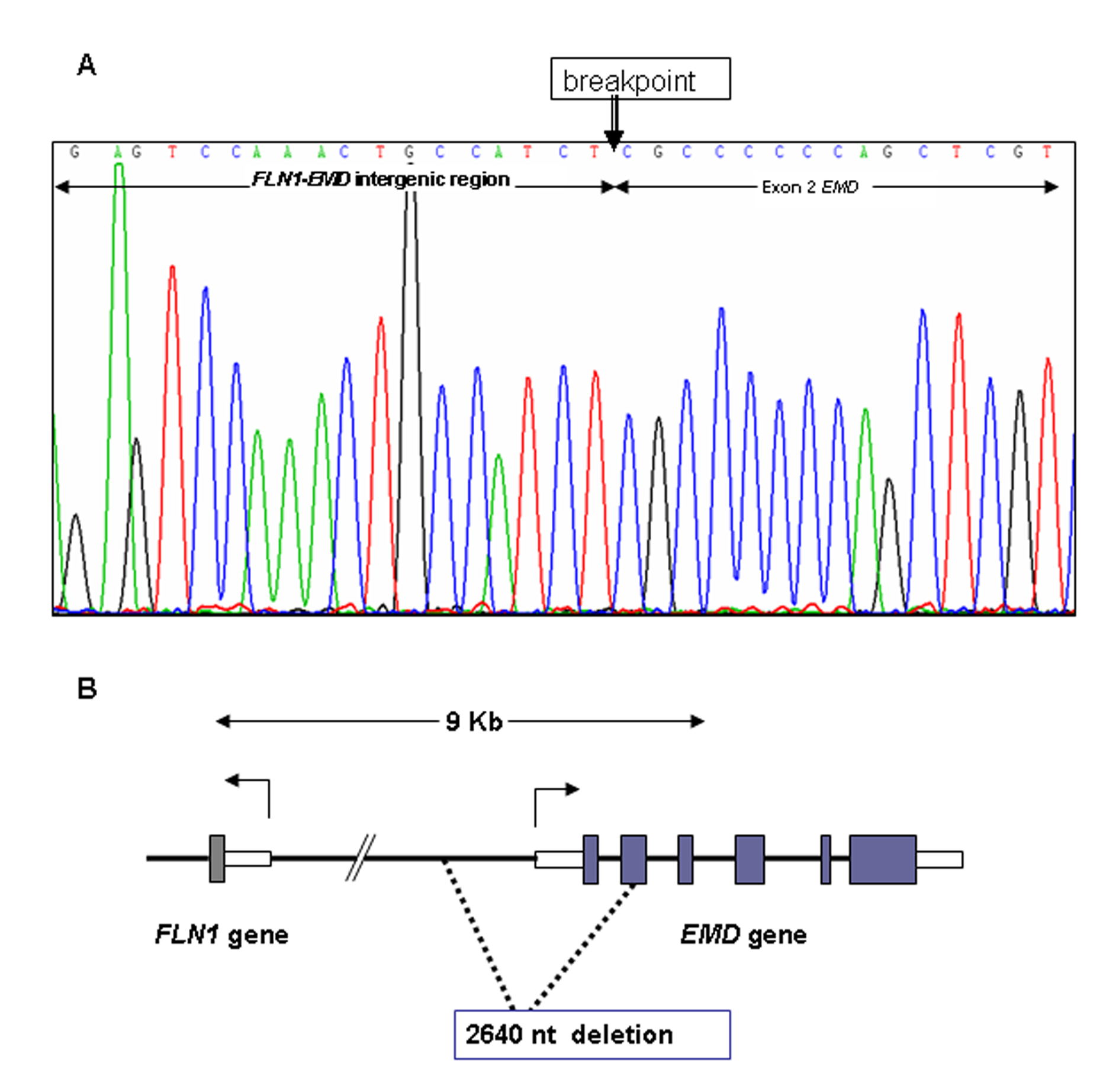 Click for large image | Figure 2. DNA sequence of the 5’ region of EMD gene from patient ED25. A) DNA sequence of the deleted allele at the breakpoint of the deletion (arrow). B) Scheme of EMD gene and deleted region of the 5’ region of EMD gene. Numbered 1-6 shaded blocks indicate exons of EMD gene. Open blocks indicate 5’-UTR and 3’-UTR of FLN1 and EMD genes. Dashed lines enclose the 2640 nucleotides deletion found in patient ED25. |
| Discussion | ▴Top |
Large emerin gene deletions are relatively rare. More than 80 mutations characterized to date are nonsense, splice site or small deletions/insertions [10].The EMD gene deletion here reported is the fifth one among the large deletions identified so far in this gene. The deletion affects the intergenic region between theEMD and FLN1 genes and extends to exon 2 of EMD gene. 5’-UTR, exon 1 and part of exon 2 of EMD gene are lost and explain the absence of emerin in the muscle. The breakpoint is more than 2 Kb far away from the 5’UTR region of the FLN1 gene. Three previously described EMD deletions, as well as the present one, involve distinct breakpoints within the 4590 bp FLN1-EMD in tergenic region. Similarly to the previously reported deletions of the EMD gene [11, 12], FLN1 gene remains intact. As suggested by Eksioglu et al [13], probably only DNA rearrangements maintaining FLN1 function are viable.
In two previous patients with a complete loss of EMD gene [11], cardiomyopathy, present as bradychardia, was treated by the insertion of a pacemaker at age 9 in one patient, whereas in other patient, cardiomyopathy was apparent as a low heart rate at age 26. In the patient here presented, the clinical course was as expected for an Emery-Dreifuss dystrophy, although the cardiac symptoms appeared much earlier than average and the patient required placement of a dual chamber transvenous pacemaker (DDDR type). A similar case with early cardiac symptoms has been described recently by Nigro et al [14] in a 14-year-old boy carrying a nonsense mutation in exon 2 of the emerin gene (c.106 A > T, p.K36X) leading to a truncated protein.
Acknowledgments
This work has been supported by funds of Hospital Universitario La Paz. Thanks are due to Ruben de Sancho for his technical assistance and to Rocio Mena and Inmaculada Rueda for helping us with DNA sequencing. Thanks also to Dr Maria J Mazon for the critical reading of the manuscript.
Abbreviations
5’-UTR: 5’-Untranslated Region; CK: creatine kinase; DDDR: dual-chamber paced, dual-chamber sensed, dual response, rate modulated device; EDMD: Emery-Dreifuss muscular dystrophy; MLPA: Multiplex Ligation-dependent Probe Amplification; PCR: DNA polymerase chain reaction
| References | ▴Top |
- Manilal S, Recan D, Sewry CA, Hoeltzenbein M, Llense S, Leturcq F, Deburgrave N, et al. Mutations in Emery-Dreifuss muscular dystrophy and their effects on emerin protein expression. Hum Mol Genet. 1998;7(5):855-864.
pubmed doi - Yates JR, Bagshaw J, Aksmanovic VM, Coomber E, McMahon R, Whittaker JL, Morrison PJ, et al. Genotype-phenotype analysis in X-linked Emery-Dreifuss muscular dystrophy and identification of a missense mutation associated with a milder phenotype. Neuromuscul Disord. 1999;9(3):159-165.
pubmed doi - Quinzii CM, Vu TH, Min KC, Tanji K, Barral S, Grewal RP, Kattah A, et al. X-linked dominant scapuloperoneal myopathy is due to a mutation in the gene encoding four-and-a-half-LIM protein 1. Am J Hum Genet. 2008;82(1):208-213.
pubmed doi - Schessl J, Zou Y, McGrath MJ, Cowling BS, Maiti B, Chin SS, Sewry C, et al. Proteomic identification of FHL1 as the protein mutated in human reducing body myopathy. J Clin Invest. 2008;118(3):904-912.
pubmed - Bonne G, Mercuri E, Muchir A, Urtizberea A, Becane HM, Recan D, Merlini L, et al. Clinical and molecular genetic spectrum of autosomal dominant Emery-Dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann Neurol. 2000;48(2):170-180.
pubmed doi - Van Tintelen JP, Tio RA, Kerstjens-Frederikse WS, van Berlo JH, Boven LG, Suurmeijer AJ, White SJ, et al. Severe myocardial fibrosis caused by a deletion of the 5' end of the lamin A/C gene. J Am Coll Cardiol. 2007;49(25):2430-2439.
pubmed doi - Liang WC, Mitsuhashi H, Keduka E, Nonaka I, Noguchi S, Nishino I, Hayashi YK. TMEM43 mutations in Emery-Dreifuss muscular dystrophy-related myopathy. Ann Neurol. 2011;69(6):1005-1013.
pubmed doi - Zhang Q, Bethmann C, Worth NF, Davies JD, Wasner C, Feuer A, Ragnauth CD, et al. Nesprin-1 and -2 are involved in the pathogenesis of Emery Dreifuss muscular dystrophy and are critical for nuclear envelope integrity. Hum Mol Genet. 2007;16(23):2816-2833.
pubmed doi - Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30(12):e57.
pubmed doi - http://www.hgmd.org.
- Small K, Warren ST. Emerin deletions occurring on both Xq28 inversion backgrounds. Hum Mol Genet. 1998;7(1):135-139.
pubmed doi - Fujimoto S, Ishikawa T, Saito M, Wada Y, Wada I, Arahata K, Nonaka I. Early onset of X-linked Emery-Dreifuss muscular dystrophy in a boy with emerin gene deletion. Neuropediatrics. 1999;30(3):161-163.
pubmed doi - Eksioglu YZ, Scheffer IE, Cardenas P, Knoll J, DiMario F, Ramsby G, Berg M, et al. Periventricular heterotopia: an X-linked dominant epilepsy locus causing aberrant cerebral cortical development. Neuron. 1996;16(1):77-87.
pubmed doi - Nigro G, Russo V, Ventriglia VM, Della Cioppa N, Palladino A, Nigro V, Calabro R, et al. Early onset of cardiomyopathy and primary prevention of sudden death in X-linked Emery-Dreifuss muscular dystrophy. Neuromuscul Disord. 2010;20(3):174-177.
pubmed doi
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Neurology Research is published by Elmer Press Inc.