







| Journal of Neurology Research, ISSN 1923-2845 print, 1923-2853 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Neurol Res and Elmer Press Inc |
| Journal website http://www.neurores.org |
Review
Volume 2, Number 3, June 2012, pages 69-81
Alzheimer’s Disease: Focus on the Neuroprotective Role of Melatonin
Venkataramanujam Srinivasana, e, Edward C Lauterbachb, Asma Hayati Ahmedc, Atul Prasadd
aSri Sathya Sai Medical Educational and Research Foundation, a Medical Sciences Research Study Center, Prasanthi Nilayam, 40-Kovai Thirunahar Coimbatore-641014, Tamilnadu, India
bDepartment of Psychiatry and Behavioral Sciences, Department of Internal Medicine, Neurology Section, and the Mercer University Center for Translational Studies in Alzheimer’s, Parkinson’s, and Neurodegenerative Diseases, Mercer University, Macon, Georgia, USA
cDepartment of Physiology, School of Medical Sciences, University Sains Malaysia, Kubang Kerian, Kelantan, Malaysia
dDepartment of Neurosciences, Fortis Hospital, Vasant Kunj, 11070, New Delhi, India
eCorresponding author: Venkataramanujam Srinivasan, Sri Sathya Sai Medical Educational and Research Foundation, Prasanthi Nilayam, 40-Kovai Thirunagar, Coimbatore-641014, Tamilnadu, India
Manuscript accepted for publication June 19, 2012
Short title: Alzheimer’s Disease
doi: https://doi.org/10.4021/jnr93w
- Abstract
- Introduction
- Molecular Pathophysiology of Alzheimer’s Disease
- Mitochondrial Involvement in Alzheimer’s Disease
- Amyloid β Peptide and Mitochondrial Interaction as Triggering Events for Alzheimer’s Disease Pathogenesis
- Melatonin, Biosynthesis and Metabolism
- Melatonin in the Treatment of Alzheimer’s Disease
- Molecular Mechanisms of Melatonin’s Anti-Amyloid Actions
- Melatonin’s Neuroprotective Role for Mitochondrial Homeostasis in Alzheimer’s Disease
- Other Possible Mechanisms of Melatonin’s Action in Alzheimer’s Disease
- Sleep-Wake Rhythm and Circadian Rhythm Disturbances in Alzheimer’s Disease
- Melatonin Levels in Alzheimer’s Disease
- Melatonin’s Potential Therapeutic Value in Alzheimer’s Disease
- Use of Other Psychopharmacological Agents for Treatment of AD
- Melatonergic Agonist Ramelteon for Treatment of AD
- Conclusion
- References
| Abstract | ▴Top |
Alzheimer’s disease (AD) is characterized by a progressive loss of memory and cognitive function, and by behavioural and sleep disturbances, including insomnia. The pathophysiology of AD has been attributed to oxidative stress-induced amyloid-β protein (Aβ) deposition. Abnormal tau protein, mitochondrial dysfunction and protein hyper-phosphorylation have been demonstrated in neural tissues of AD patients. AD patients exhibit severe sleep-wake disturbances and these sleep disturbances are associated with rapid cognitive decline and memory impairment. Optimally effective management of AD patients will likely require a drug that can arrest Aβ - induced neurotoxic effects and can also restore the disturbed sleep-wake rhythm, with improvement in sleep quality. In this context, the pineal hormone melatonin has been demonstrated to be an effective antioxidant that can prevent Aβ-induced neurotoxic effects through a variety of mechanisms. Sleep deprivation itself produces many effects including oxidative damage, impaired mitochondrial function, neurodegenerative inflammation, altered proteosomal processing and abnormal activation of enzymes. In addition to treating the signs and symptoms of AD, treatment of sleep disturbances may also be necessary for preventing and arresting AD progression. Besides melatonin, a number of melatonergic agonists such as ramelteon, agomelatine and tasimelteon are now used clinically for treating insomnia and other sleep disorders. Their use may also be beneficial in treating Alzheimer’s disease.
Keywords: Alzheimer’s disease; Melatonin; Amyloid-β protein; Insomnia; Anti-oxidant; Sleep
| Introduction | ▴Top |
Age-associated neurodegenerative diseases like Alzheimer’s disease (AD) and Parkinson’s disease (PD) are attributed to increased oxidative damage of the central nervous system [1-5]. Because of its high rate of oxygen consumption and high content of polyunsaturated fatty acids, the brain is easily susceptible to increased oxidative stress. Increased protein, lipid and DNA oxidation has been demonstrated in brain samples of AD patients [6]. Protein carbonyl 3, 3’-dityrosine and 3-nitrotyrosine levels in post-mortem brain samples of AD patients exhibit increased oxidative and nitrosative protein modification in the hippocampal and neocortical regions [7].
Oxidative damage is related to other primary cytopathological features of AD, including mitochondrial demise and amyloid-β (Aβ) protein. In addition to age, the other major risk factor for AD is gender. The incidence of AD is higher in women than in men, and this has been attributed to increased mitochondrial toxicity induced by Aβ proteins [8]. Increased Aβ levels trigger all of the most important pathological features of AD, including tau hyper-phosphorylation, formation of neurofibrillary tangles, synaptic dysfunction, and neuronal cell death [9]. Abnormal accumulation of an Aβ protein results in the formation of toxic oligomers that cause synaptic damage, alterations in glutamate receptors, circuit hyperexcitability and alterations in signalling pathways related to synaptic plasticity [10]. Recent studies implicate mitochondrial pathology as an important contributory factor in the late onset forms of AD [11].
Many of the current drugs used in the treatment of AD improve symptoms but do not have any significant disease-modifying effects. It is estimated that the cost of care to the average AD patient family exceeds $ 90,000 per year [12], while the societal cost exceeds $ 100 billion per annum [13, 14]. From these figures it is evident that there is an urgent need for speedy implementation of preventive treatment programs for arresting the progress of this disease and other neurodegenerative diseases. In the last 15 years, rapid developments have taken place on the role of the pineal hormone melatonin in arresting the neurotoxic effects induced by Aβ in animal studies. Melatonin has been shown to antagonize Aβ-induced effects such as increased oxidative stress, increased lipid peroxidation, formation of protein carbonyl products, and mitochondrial pathological mechanisms [15]. Thus, melatonin has substantial potential to prevent or delay AD progression, thereby reducing patient symptomatic burden, family expense, and societal cost. Additionally, its capacity to improve sleep disorders can improve AD symptoms and potentially reduce AD neuropathology, as discussed below.
| Molecular Pathophysiology of Alzheimer’s Disease | ▴Top |
In Alzheimer’s disease there is progressive loss of cognitive function, with other neurobehavioral abnormalities like agitated behaviour and profound sleep disturbances.
Despite volumes of studies undertaken on AD, its etiology remains unknown. Many mechanisms including chronic inflammation associated with cytokine release, trace element neurotoxicity and free radical generation have each been suggested as possible contributory factors underlying the etiology of AD. The deposition of amyloid plaques in AD causes cell death by induction of oxidative stress, a primary pathogenic mechanism of AD. Aβ protein deposition initiates flavo-enzyme dependent increases in H2O2 and lipid peroxides that increase free radical generation [16]. This increased β-amyloid protein - induced oxidative stress in conjunction with decreased neurotrophic support [17] are the major determinants of AD. Studies undertaken in post-mortem brain samples obtained from AD patients have shown extensive lipid, protein and DNA oxidation [7]. Neural tissues of AD patients exhibit increased levels of peroxidation end-products such as malondialdehyde (MDA), 4-hydroxynonenal, carbonyls and other species [18]. Though Aβ contributes directly or indirectly to neuronal degeneration, its potential to cause AD depends on an individual’s susceptibility to Aβ-mediated toxicity.
| Mitochondrial Involvement in Alzheimer’s Disease | ▴Top |
Mitochondria are not only the major source of reactive oxygen species (ROS) but also the primary target of attack by ROS and reactive nitrogen species [19]. Damage to the mitochondrial respiratory chain can cause breakdown of the mitochondrial membrane proton potential, opening of the mitochondrial permeability transition pore (mtPTP) and consequent induction of apoptosis, leading to further generation of free radicals and maintaining a vicious cycle that ultimately results in cell death by either necrotic or apoptotic processes [20]. Findings from a number of studies reveal the involvement of mitochondrial ROS production and subsequent mitochondrial abnormalities in the pathophysiology of AD [21]. Mitochondrial superoxide (O2-) production plays a critical role in the pathological events that follow elevations of Aβ peptide levels. The histopathological changes of AD, namely extracellular accumulation of oligomeric and fibrillar Aβ peptides and intracellular neurofibrillary tangles induce functional deficits of the mitochondrial respiratory chain complexes and, thereby, result in mitochondrial dysfunction and enhanced oxidative stress [22].
| Amyloid β Peptide and Mitochondrial Interaction as Triggering Events for Alzheimer’s Disease Pathogenesis | ▴Top |
The amyloid cascade hypothesis proposed by Hardy and Selkoe (2002) suggests the faulty metabolism of amyloid precursor protein as the initiating event in the pathogenesis of AD [23]. Whether extracellular Aβ - induced mitochondrial damage is the initial triggering event in AD is a subject of debate since Aβ appears within the mitochondria long before the appearance of extracellular Aβ deposits. The enzymes that transfer Aβ to mitochondria have been identified as a complex of translocases of both the outer membrane (TOM) and inner membrane (TIM) [24]. The intracellular presence of Aβ is one of the major reasons for the reduction of oxygen consumption caused by the electron transport chain, since Aβ diminishes the enzymatic activity of respiratory chain complexes III and IV [23]. Several other neurotoxic mechanisms, such as the formation of ionic channels that allows increased calcium uptake by mitochondria and mtPTP opening with subsequent inhibition of respiratory complexes [25], have been proposed for Aβ - induced neurotoxicity that is mediated through intramitochondrial interactions. The fact that Aβ can accumulate both intracellularly and intramitochondrially has been demonstrated by Rosales-Corral et al (2012) [26], in which intracerebral injection of fibrillar Aβ caused accumulation of Aβ both intracellularly and intramitochondrially, deep within the cristae, thereby supporting other investigators’ views of intramitochondrial accumulation of Aβ. In addition to inhibiting mitochondrial respiratory complex activities in AD patients, disturbances in mitochondrial dynamics have also been demonstrated. This includes disturbances in mitochondrial structure, impairment of dynamin - related peptide (Drp1) and imbalances in fission and fusion, with consequent neuronal damage and synaptic loss [27]. Significant disturbances of mitochondrial proteins and lipids also occur following intramitochondrial accumulation of Aβ, which in turn causes functional impairment of mitochondria in AD [26].
Interaction of Aβ with cyclophilin D (CypD), a Ca2+-associated protein found in the mtPTP, causes CypD translocation from the matrix to the inner membrane, resulting in opening of the mtPTP and consequent mitochondrial swelling, with ensuing cellular and synaptic alterations [28]. CypD deficiency has been shown to attenuate Aβ-induced mitochondrial oxidative stress, thereby reducing Aβ-related synaptic dysfunction and cognitive impairment. From this finding it is clear that the interaction of Aβ and Cyp D is one of the major reasons for mitochondrial pathology in AD. Thus, the mitochondrial aspect of AD with regard to Aβ-induced free radical generation, excitation-dependent calcium overload and its consequences for the mitochondrial membrane potential and mtPTP all must be taken into account to effectively tackle the disease with suitable drug therapy. Attenuation or prevention of increased oxidative stress seen in AD patients should be the primary goal of strategic treatment for AD.
| Melatonin, Biosynthesis and Metabolism | ▴Top |
Melatonin (N-acetyl-5-methoxytryptamine) is synthesized mainly by the pineal gland in all mammals but is also formed by many other organs and tissues in the body. It is synthesized from serotonin, and therefore ultimately from tryptophan. Serotonin is acetylated to form N-acetylserotonin by the rate limiting enzyme arlakylamine-N-acetyltransferase (AA-NAT). N-acetylserotonin is then converted into melatonin by the enzyme hydroxyl-indole-O-methyl transferase. The biosynthetic pathway is given in Figure 1. Once formed, melatonin is not stored within the pineal gland but diffuses into the capillary blood and CSF. As melatonin passes through all tissues with ease and CSF melatonin values are nearly 30 times higher than those in the blood, brain tissue has higher melatonin concentrations than any other tissue in the body [29]. While tissue melatonin only exhibits a moderate circadian variation, circulating melatonin exhibits most pronounced circadian rhythms, with the highest levels occurring at night and the lowest levels during daytime in most of the age groups (10 - 70yrs).
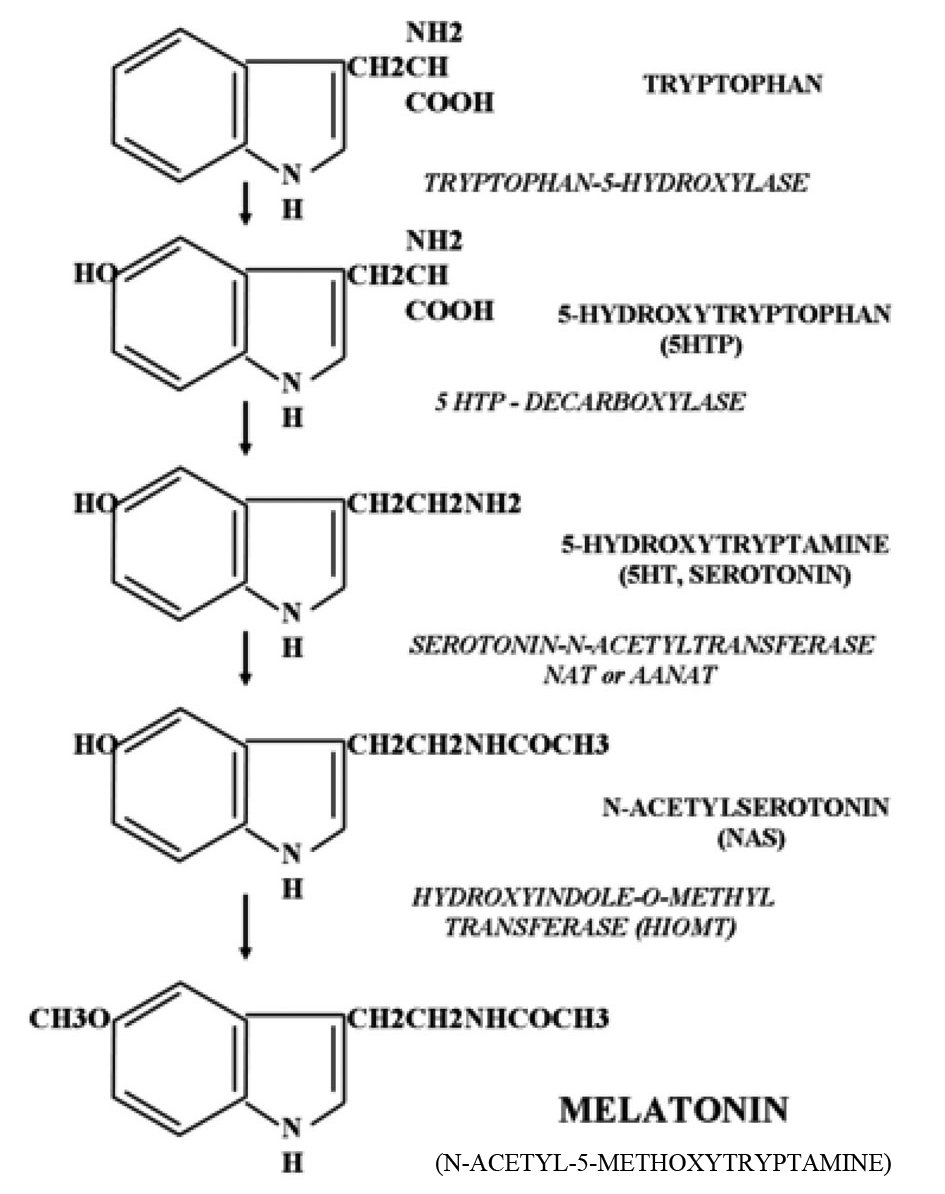 Click for large image | Figure 1. Biosynthesis of melatonin. |
Circulating melatonin is metabolized mainly in the liver where it is first hydroxylated at the C6 position by cytochrome P450 mono-oxygenases (CYP1A1 and CYP1A2) and, thereafter, conjugated with sulphate to form 6-sulfatoxymelatonin (aMT6S) [30]. In the brain, melatonin is metabolized to kynuramine derivatives N1-acetyl-N2-formyl-5-methoxykynuramine (AFMK) [31]. A schematic diagram showing the metabolism of melatonin is provided in Figure 2.
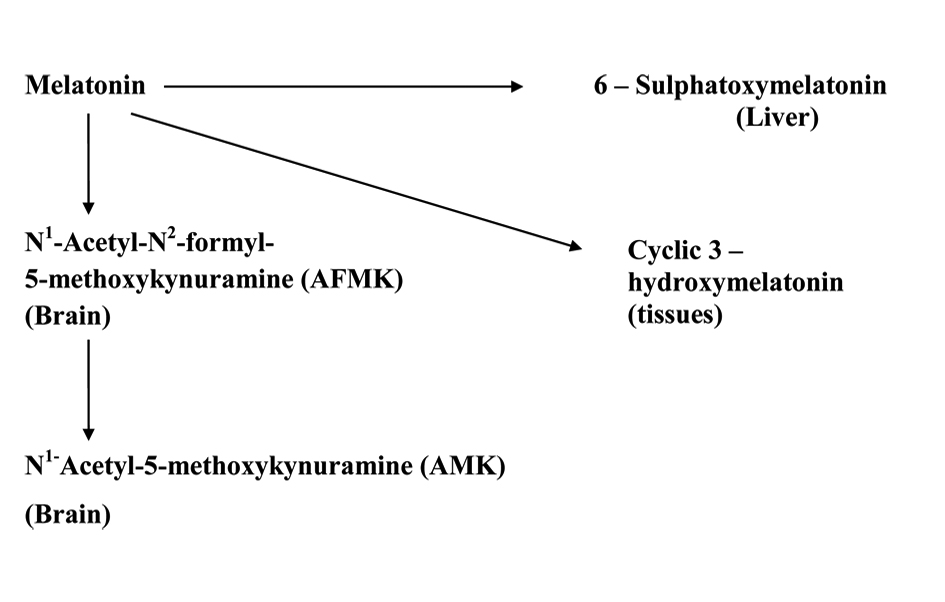 Click for large image | Figure 2. Schematic diagram showing melatonin’s metabolic pathways in periphery and in CNS. |
Melatonin is involved in the control of various physiological functions of the body, such as co-ordination of circadian rhythms including sleep-wakefulness rhythms, sleep regulation, immune function, anti-oxidant functions, control of reproduction, inhibition of tumor growth, and the control of human mood and behaviour and these have been reviewed [32].
Melatonin participates in many of these functions by acting through G-protein membrane receptors like MT1 and MT2 melatonin receptors [33]. Melatonin also acts directly on cells without the intervention of any of these receptors by binding to calmodulin [34]. Although the free radical scavenging action of melatonin does not involve any receptors, melatonin‘s stimulatory action on the formation of the anti-oxidant enzyme γ-glutamylcysteine synthase involves RZR/ROR α receptors [35].
| Melatonin in the Treatment of Alzheimer’s Disease | ▴Top |
Melatonin is an interesting neuroprotective agent as it displays multiple properties through which it antagonizes oxidative stress. One might classify the effects of melatonin as: 1) antioxidant, including influences on mitochondrial metabolism; 2) antifibrillogenic; 3) cytoskeletal, including the suppression of protein hyperphosphorylation. Although these actions are observed at pharmacological concentrations, the relevance of these findings can be appreciated if one considers the relatively high rates of melatonin secretion into the CSF, uptake into the brain tissue and metabolization of melatonin into other neuroprotective compounds such as the kynuramines AFMK and AMK [36]. Melatonin has been shown to prevent the death of neuroblastoma cells exposed to β-amyloid polypeptide. This was established by the pioneering work of Pappolla’s research group. Using murine neuroblastoma cells (N2a), Pappola et al, 1997 [37, 38] first demonstrated that co-incubation of neuroblastoma cells with amyloid-β polypeptide and melatonin significantly reduced several features of apoptosis, including cellular shrinkage and formation of membrane blebs.
Recent studies of astrocytes show that astrocytic apoptosis contributes to the pathogenesis of AD. Astrocytes exhibit tau phosphorylation and activation of stress kinases, as seen in AD neuronal pathology. They also produce apolipoprotein-E4 (apoE4), aggravating β-amyloid effects [39]. The β-amyloid protein and astrocyte–neuron interaction potentiates the neurodegeneration of AD. During interaction with Aβ, astrocytes lose control over glial NO production, thereby forming neurotoxic peroxynitrate. Treatment of C6 astroglioma cells with melatonin effectively prevented the increase in NO production induced by Aβ [40].
Several animal models of AD have been used to study the possible antioxidative and antiapoptotic actions of melatonin in arresting neuronal lesions. Okadaic acid induces physiological and biochemical changes similar to those seen in AD. Increased levels of 4-hydroxynonenal (a product of lipid peroxidation) in cultured neuronal cells (SY5Y cells) has been found following administration of okadaic acid [41]. With administration of antioxidants like vitamin C or melatonin, the effects of okadaic acid on NIE 115 neuronal cells was effectively prevented [42]. Melatonin was found superior to vitamin C in this study, since it not only prevented free radical-induced damage with greater efficiency but also increased the enzymes glutathione-S transferase and glutathione reductase, which was not evident with vitamin C [42]. In a model of APP695Tg mice, senile plaques appear in the cerebral cortex as early as at 8 months of age. These mice display behavioural manifestations and memory impairments as seen in AD patients. Administration of melatonin (10 mg/kg) alleviated learning and memory deficits and also reduced the number of apoptotic neurons [43].
| Molecular Mechanisms of Melatonin’s Anti-Amyloid Actions | ▴Top |
Melatonin has not only reduced several features of apoptosis, like cellular shrinkage, or formation of membrane blebs as discussed in the earlier paragraphs, but also exerts its antiamyloid actions through several mechanisms. Aβ by inducing oxidative stress damages mitochondrial DNA, forms protein carbonyl, induces lipid peroxidation and impairs the mitochondrial membrane structure and respiration and breaks down the mitochondrial membrane potential. Each of these actions that produce mitochondrial dysfunction was prevented by melatonin administration. Melatonin also inhibits the formation of amyloid fibrils, as demonstrated by different techniques. Both Aβ1-40, and Aβ1-42 peptides were effectively inhibited [44-45]. A structural analog of melatonin, namely indole-3-propionic acid, not only shares melatonin’s radical scavenging activity [46] but also exhibits similar or even higher antifibrillogenic activity [45-46]. Lipoproteins including cholesterol and apolipoprotein subtypes can modulate fibrillogenesis. Melatonin has been shown to reverse the profibrillogenic activity of apolipoprotein E4 and to antagonize neurotoxic combinations of Aβ and apoE4 or apoE3 [47]. One other manifestation of AD as studied in experimental models of AD is the expression of protein hyper phosphosphorylation and cytoskeletal disorganization driven by glycogen synthase kinase 3 (GSK-3), but melatonin also reversed GSK-3 activation, showing thereby that melatonin not only acts as an antioxidant but also interferes with the phosphorylation system, especially stress kinases [48]. Melatonin has attenuated tau hyperphosphorylation induced by wortmannin [49]. Tyrosine kinase (trk) receptors [50], important elements of the phosphorylation system and neurotrophins, are adversely affected by Aβ and other oxidotoxins. In neuroblastoma cells, melatonin normalized trk and neurotrophin expression [50]. A schematic diagram showing melatonin’s neuroprotective actions is presented in Table 1 and Figure 3.
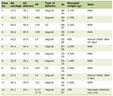 Click to view | Table 1. Summary of Melatonin’s Neuroprotective Effects in AD: Mechanisms |
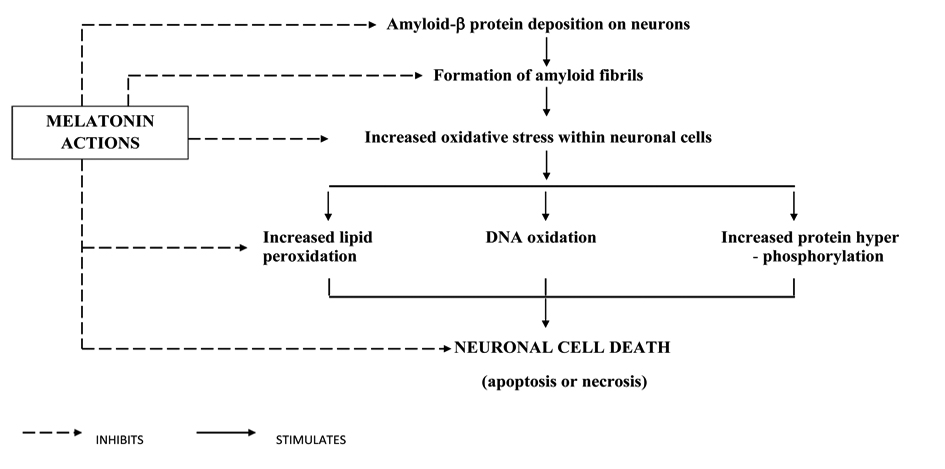 Click for large image | Figure 3. Pathophysiology of AD and melatonin’s neuroprotective actions. |
| Melatonin’s Neuroprotective Role for Mitochondrial Homeostasis in Alzheimer’s Disease | ▴Top |
Mitochondrial abnormalities in AD have been discussed in the earlier paragraphs. Melatonin’s neuroprotective role in AD not only depends upon its radical scavenging actions but also on its additional actions, including protecting the mitochondrial membranes and mitochondrial DNA from oxidative insults, stimulation of glutathione (GSH) synthesis, reduction of oxidized glutathione (GSSG) levels and maintenance of mitochondrial electron flux [20]. The electron transport chain (ETC) represents a major source of reactive oxygen species (ROS) within the cell [51]. Complex I and III of the ETC have been identified as the two principal sites of superoxide anion (O2-) generation [52]. While much of the O2- is released from complex III to either side of the inner membrane, the iron-sulfur cluster N2 of complex I appears to be the main site of O2- release to the matrix [53]. The O2- so formed is disposed of by several pathways. A portion of it re-donates electrons to the ETC at cytochrome c [54]. Another fraction is converted into H2O2 and O2 by the mitochondrial Mn-containing subform of super oxide dismutase (MnSOD) [55]. A third fraction of O2-combines with NO to give rise to peroxynitrite, an additional source of destructive proteins. Melatonin’s mitochondrial actions take place within the organelle. Melatonin, possessing a balanced amphilicity, crosses the cell membranes with ease and is concentrated within subcellular compartments [56]. Melatonin’s effects on electron flux have two aspects. First, it increases the activities of mitochondrial respiratory complexes I and IV in a time dependent manner [57]. This effect of melatonin, namely the improvement of electron transport capacity in mitochondria, is remarkable as these effects are observed in senescence-accelerated mice [58]. In addition to this, other processes perturbing the mitochondrial membrane potential such as calcium overload are also antagonized by melatonin. Melatonin has been shown to prevent calcium overload and counteracted the collapse of the mitochondrial membrane potential induced by H2O2 [59], and also inhibited the opening of the mtPTP, thereby preventing the occurrence of apoptosis. A summary of melatonin’s neuroprotective actions on mitochondrial physiology and metabolism are presented in Table 2 and Figure 4. Melatonin also activates signalling pathways, namely the Bcl-2 pathway that stabilizes mitochondrial function by antiapoptotic Bcl-2 family modulators. Melatonin enhanced Bcl-2 expression with consequent inhibition of Aβ-induced cell death [60]. Caspase-3 is known to be directly linked to cytochrome c release and is related to cell death in AD [61, 62], and this is down regulated by melatonin [63]. Thus, the apoptosis-inducing factors liberated during mitochondrial damage are effectively inhibited by melatonin through a variety of mechanisms, thereby preventing the neuronal cell death seen in AD.
Click to view | Table 2. Summary of Melatonin’s Beneficial Physiologic and Metabolic Effects on Mitochondrial Dysfunction Induced by Amyloid-Β in Neural Tissues |
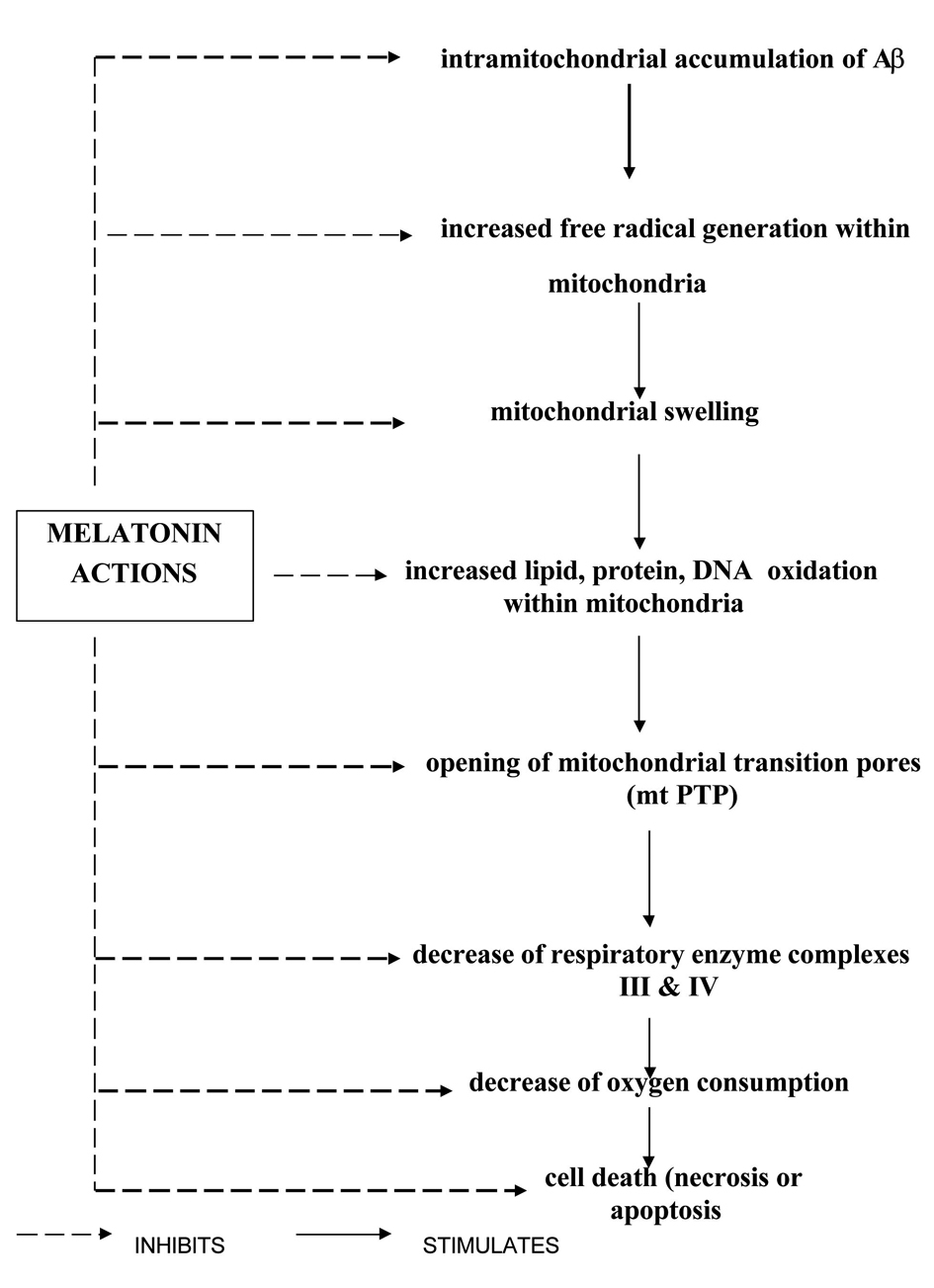 Click for large image | Figure 4. Melatonin’s effects on mitochondrial pathophysiology in AD. |
| Other Possible Mechanisms of Melatonin’s Action in Alzheimer’s Disease | ▴Top |
Glutamate receptors, particularly NMDA receptors, are involved in the pathophysiology of AD [64]. Excessive glutamate activity resulting in excessive calcium influx activates a number of enzymes including phospholipases, endonucleases, neuronal nitric oxide synthase and proteases that can result in neuronal toxicity leading to several neurodegerative diseases including AD [64]. Melatonin, by reducing the NMDA-induced high Ca2+ influx, thereby offers a neuroprotective effect. The regulation of intracellular Ca2+ by melatonin is explained through its action on melatonin MT2 receptors. cAMP-dependent protein kinase (PKA) is involved in the activation of calcium channels [64]. Melatonin, by acting through MT2 receptors, decreases cAMP formation and thereby blocks PKA activation [65]. Findings in AD patients reveal that both MT1 and MT2 receptors are decreased in the cortical regions of AD patients [66].
| Sleep-Wake Rhythm and Circadian Rhythm Disturbances in Alzheimer’s Disease | ▴Top |
A number of studies in AD patients have shown that there are profound disturbances in the sleep-wake cycle correlating with the progression of the disease. Cross-sectional studies reveal that sleep disturbances are associated with memory and cognitive impairment. Clinical studies in AD patients favour disruption of the circadian timing system in AD since numerous overt rhythms including body temperature, glucocorticoid, melatonin and other hormonal rhythms are disturbed [67-69]. Phase differences between the rest-activity and core body temperature cycles are significantly delayed in AD patients [70]. A chronobiological phenomenon observed in AD patients in conjunction with sleep-wake disturbances is “sun downing,” in which symptoms such as disorganized thinking, reduced ability to maintain attention on external stimuli, and motor disturbances like agitation, wandering, perceptual and emotional disturbances all tend to appear in the late afternoon or early evening [71]. Chronotherapeutic procedures including exposure to bright light in selected circadian phases alleviated sundowning symptoms like wandering, agitation and delirium and also improved sleep-wake patterns in AD patients [72].
| Melatonin Levels in Alzheimer’s Disease | ▴Top |
Studies of melatonin levels in AD patients reveal that they are lower in AD patients compared to age matched controls [73-76]. Decreased CSF melatonin levels in AD patients were attributed to decreased melatonin production rather than to diluting effects of CSF. CSF melatonin levels decreased even in preclinical stages (Braak stage-1) when patients did not yet manifest cognitive impairment, suggesting that a reduction in CSF melatonin may be an early marker for the first stages of AD [76, 77]. The decrease of melatonin levels in AD has also been attributed to defective retino-hypothalamic tract or suprachiasmatic nucleus-pineal connections [78]. However, decreased nocturnal melatonin levels have also been shown to correlate with the severity of mental impairment of patients with dementia [78]. Recently it has been suggested that the choroid plexus portal theory of CSF circulation offers an explanation for the neuropathology of AD. Contrary to earlier views expressed in medical text books that the cerebrospinal fluid absorption occurs through the arachnoid granules into the superior sagittal sinus, present evidence indicates that cerebrospinal fluid is moved from the choroid fissure into the ventricular system. Understanding the circulation of CSF through these portals, combined with understanding of the consequences of deficient CSF melatonin, may provide answers to the puzzles of AD pathophysiology [79].
| Melatonin’s Potential Therapeutic Value in Alzheimer’s Disease | ▴Top |
As AD patients have a profound deficiency in endogenous melatonin production, replacement of physiological levels of melatonin in the brain may represent a kind of treatment for arresting the progress of this disease. Melatonin’s vasoprotective properties could conceivably help in enhancing cerebral blood flow and could therefore help to improve the clinical condition of AD patients. Moreover AD patients show a greater breakdown of the circadian sleep-wake cycle when compared to similarly aged non-demented controls. As sleep disturbances exacerbate memory and cognitive impairment, amelioration of sleep disturbances is of paramount importance in treating AD patients [80]. In a study conducted in 14 AD patients with 6 - 9 mg of melatonin/day and follow up over a 2 - 3 year period, it was noted that melatonin improved sleep quality in most of the patients [81, 82].
Sundowning, diagnosed clinically, was no longer detectable in 12 out of 14 patients. Reduction in cognitive impairment and amnesia was noted. This should be contrasted with significant deterioration of the clinical conditions of the disease expected in patients after a 1 - 3 year evolution of AD [83]. The efficacy of melatonin in treating AD patients is supported by a number of other studies. Administration of melatonin (6 mg/day) for 4 weeks to 7 AD patients reduced night-time activity compared to placebo [83]. The improvement of sleep and alleviation of sundowning was reported in 11 elderly AD patients treated with melatonin (3 mg/day at bedtime) [72]. Significant decreases in agitated behaviour and daytime sleepiness were also noted in this study. Reduction of sun downing with the use of 3 mg of melatonin was reported using actigraphy in 7 AD patients [84]. In a double blind study conducted on AD patients, it was noted that 3 mg/day of melatonin significantly prolonged actigraphically evaluated sleep time, decreased activity at night-time and improved cognitive functions [85]. In a multicenter, randomized, placebo-controlled clinical trial in a large sample of 157 AD patients with sleep disturbances, patients were randomly assigned to one of three treatment groups (placebo, 2.5 mg slow release melatonin or 10 mg melatonin). Melatonin or placebo was administered for a period of 2 months with actigraphic monitoring, and an increased nocturnal total sleep time and decreased waking after sleep onset was noted in the melatonin group. On subjective measures using caregiver ratings, significant improvement in sleep quality was noted in the 2.5 mg sustained release melatonin group relative to the placebo group [86]. However in a recent study, melatonin 8.5 mg immediate release and 1.5 mg sustained release was administered at 10:00 PM for 10 consecutive nights to patients with Alzheimer’s disease and no significant difference was noticed between placebo and melatonin on sleep, circadian rhythms or agitation [87]. The lack of beneficial effect of melatonin on sleep was attributed to supraphysiological doses of melatonin. Besides tackling the sleep and circadian rhythm disturbances seen in AD, melatonin can also act at multiple different levels in AD. The antioxidant, mitochondrial and antiamyloidogenic effects of melatonin can be viewed as potentially interfering with the onset of the disease. As such, melatonin has several obvious advantages over other comparable compounds, including most other antioxidants. The question of whether melatonin has therapeutic value in preventing or treating AD, affecting disease initiation or progression of the neuropathology and its driving mechanisms remains to be answered by future multicenter double-blind clinical trials. In the meantime, it is suggested that improvement of insomnia in neurodegenerative conditions and particularly in AD with melatonin represents a clinical choice that may be of help in arresting the progression of the disease as well as offering neuroprotection against the development of AD.
| Use of Other Psychopharmacological Agents for Treatment of AD | ▴Top |
In a recent review of psychopharmacological approaches to neuroprotection, one of the authors (ECL) suggested a trial of lithium (a mood stabilizer) with melatonin since this combination can potentially synergize neuroprotective effects on Aβ, hyperphosphorylated tau, ROS, mtPTP, and apoptosis, with lithium also potentially improving ubiquitylation [88]. It is suggested that treatments lowering plasma Aβwill be beneficial for reducing the risk of developing AD.
| Melatonergic Agonist Ramelteon for Treatment of AD | ▴Top |
Ramelteon is (s)-N-(2-(1,6,7,8-tetrahydro-2H-indeno(5,4-b)furan-8-yl)ethyl)propionamide. This melatonin receptor agonist has a chemical formula C16H21NO2 with a molecular weight of 259.34. Its affinity for MT1 and MT2 receptors is highly selective, with little affinity to quinine reductase (formerly known as MT3receptors) [89].
The selectivity of ramelteon for MT1 receptors is 1000-fold more than its affinity to MT2 receptors. Ramelteon is metabolized in the liver via oxidation to hydroxyl and carbonyl groups and then conjugated with glucuronide. CYPA2 is the major hepatic enzyme involved in ramelteon metabolism. Four principal metabolites of ramelteon, namely M-I, M-II, M-III and M-IV have been identified [90]. Among the M-II occurs at a high concentration with its systemic level being 20 to 100-fold greater than ramelteon. A number of clinical studies of the use of ramelteon in primary insomnia in both adult and elderly patients have shown that it is very effective in reducing sleep onset latency and increasing total sleep time and sleep efficiency. Studies on the efficacy of ramelteon in treating insomnias have been published elsewhere [91-93]. As ramelteon has greater potency and affinity at melatonin receptors, it is suggested that melatonin’s neuroprotective effects could be shared by ramelteon with greater efficacy and can be of value in treating Alzheimer’s disease [94]. The neuroprotective effect of ramelteon in AD is inferred from the recent finding that ramelteon increased the neural content of BDNF in cultured mouse cerebellar granule cells [95]. Since neither melatonin nor its agonist ramelteon has undergone clinical trials for clinical and disease modifying neuroprotective properties in neurodegenerative diseases, ramelteon’s clinical findings remain to be established in neurodegenerative disease patients [94]. Melatonin and ramelteon, because of their sleep promoting actions, may have an important role to play in the treatment of Alzheimer’s disease and other neurodegenerative diseases. Of these, melatonin may be more significant because of its probable involvement in the aetiology of AD itself. Deficiency of CSF melatonin is postulated as one of the major reasons for AD neuropathology as pointed out in the earlier paragraphs.
The loss of this antioxidant in the CSF will expose vital neurons to increased oxidative stress with resultant cell destruction and the development of AD [79]. A year’s melatonin intake at a dose of 9 mg/day will cost only $ 25 and could greatly benefit the patient in slowing the progression of AD [79].
| Conclusion | ▴Top |
Although the exact cause for AD is still under intense investigation, available evidence strongly implies the deposition of amyloid-β protein and oxidative stress and their neurotoxic effects as the major contributing factors to the pathogenesis of AD. Intramitochondrial accumulation of amyloid-β protein is one of the major reasons for mitochondrial pathophysiology in AD. Inhibition of respiratory enzyme complexes, with increased electron leakage, decreased oxygen consumption and decreased ATP synthesis caused by increased oxidative stress ultimately contribute to neuronal cell death by necrosis or apoptosis. AD patients have profound sleep-wake cycle disturbances. The hormone melatonin, involved in the regulation of sleep and circadian rhythms, is greatly diminished in patients with AD. Melatonin also has been demonstrated as an effective antioxidant both in vivo and in vitro model systems. Experimental studies using neuronal cell cultures or animal models of AD have shown that melatonin has been effective in preventing Aβ-induced toxic effects on neuronal cells. Melatonin also exerts its beneficial effects intramitochondrially, increasing respiratory enzyme complex activities, increasing the flow of electrons, scavenging free radicals and preventing oxidative stress-induced lipid, protein and DNA oxidation within cells and also within mitochondria. Treatment of AD patients with melatonin has clearly improved sleep quality in almost all studies undertaken and it also has been beneficial in improving cognitive functions. The preventive potential of melatonin in AD disease progression remains to be established by clinical trials. In this respect, the effect of a newly introduced sleep promoting drug, namely ramelteon, a melatonin agonist, may be of benefit since it can improve both sleep quality and may also offer neuroprotection against pathogenic proteins, free radical-induced lipid peroxidation and mitochondrial transition pore development. Multicenter placebo controlled clinical trials using ramelteon have to be undertaken to prove the efficacy of this drug in preventing or arresting the progression of AD.
Conflict of Interest
All authors of this paper declared that they have no conflict of interest. V. Srinivasan (the corresponding author) has not received any pay or salary duringthe last three years and has published nearly twenty review/research papers in international medical journals with an international collaborative team. Sri Sathya Sai Medical Educational and Research Foundation, A Medical Sciences Research Study Centre (founded by DR. V. Srinivasan, PhD., MAMS (Member, National academy of Medical Sciences India), has not received any research grant from any Governmental, Private, Public, National or International Funding agencies, since its inception in 2009.
| References | ▴Top |
- Vollicer L, Crino B. Involvement of free radicals in dementia of Alzheimer’s type hypothesis. Neurobiol Aging 1990;11:567-571.
- Markesbery WR. Oxidative stress hypothesis in Alzheimer's disease. Free Radic Biol Med. 1997;23(1):134-147.
pubmed doi - Christen Y. Oxidative stress and Alzheimer disease. Am J Clin Nutr. 2000;71(2):621S-629S.
pubmed - Smith MA, Rottkamp CA, Nunomura A, Raina AK, Perry G. Oxidative stress in Alzheimer's disease. Biochim Biophys Acta. 2000;1502(1):139-144.
pubmed - Srinivasan V. Melatonin oxidative stress and neurodegenerative diseases. Indian J Exp Biol. 2002;40(6):668-679.
pubmed - Subbarao KV, Richardson JS, Ang LC. Autopsy samples of Alzheimer's cortex show increased peroxidation in vitro. J Neurochem. 1990;55(1):342-345.
pubmed doi - Pamplona R, Dalfo E, Ayala V, Bellmunt MJ, Prat J, Ferrer I, Portero-Otin M. Proteins in human brain cortex are modified by oxidation, glycoxidation, and lipoxidation. Effects of Alzheimer disease and identification of lipoxidation targets. J Biol Chem. 2005;280(22):21522-21530.
pubmed doi - Vina J, Lloret A. Why women have more Alzheimer's disease than men: gender and mitochondrial toxicity of amyloid-beta peptide. J Alzheimers Dis. 2010;20 Suppl 2:S527-533.
pubmed - Simon AM, Frechilla D, del Rio J. [Perspectives on the amyloid cascade hypothesis of Alzheimer's disease]. Rev Neurol. 2010;50(11):667-675.
pubmed - Crews L, Masliah E. Molecular mechanisms of neurodegeneration in Alzheimer's disease. Hum Mol Genet. 2010;19(R1):R12-20.
pubmed doi - Swerdlow RH, Redpath GT, Binder DR, Davis JN, 2nd, VandenBerg SR.Mitochondrial DNA depletion analysis by pseudogene ratioing. J Neurosci Methods. 2006;150(2):265-271.
pubmed doi - Aarsland D, Sharp S, Ballard C. Psychiatric and behavioral symptoms in Alzheimer's disease and other dementias: etiology and management. Curr Neurol Neurosci Rep. 2005;5(5):345-354.
pubmed doi - Alzheimer’s Association: Alzheimer’s disease Facts and Figures, Chicago, Alzheimer’s Association, 2007.
- Rice DP, Fillit HM, Max W, Knopman DS, Lloyd JR, Duttagupta S. Prevalence, costs, and treatment of Alzheimer's disease and related dementia: a managed care perspective. Am J Manag Care. 2001;7(8):809-818.
pubmed - Pappolla MA, Chyan Y-J, Bozner P, Soto C, Reiter RJ, Brewer G, Robakis NK, Zagorski MG, Frangione B, Ghiso J. Dual anti-amyoidogenic and anti-oxidant properties of melatonin. A new therapy for Alzheimer’s Disease. In Research Advance in Alzheimer’s disease Eds Iqbal K, Mortimer JA, Winblad B, Wisniewski HM, New York ,Wiley, 1999, pp 661-669.
- Hensley K, Carney JM, Mattson MP, Aksenova M, Harris M, Wu JF, Floyd RA, et al. A model for beta-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer disease. Proc Natl Acad Sci U S A. 1994;91(8):3270-3274.
pubmed doi - Hock C, Heese K, Muller-Spahn F, Hulette C, Rosenberg C, Otten U. Decreased trkA neurotrophin receptor expression in the parietal cortex of patients with Alzheimer's disease. Neurosci Lett. 1998;241(2-3):151-154.
pubmed doi - Markesbery WR, Carney JM. Oxidative alterations in Alzheimer's disease. Brain Pathol. 1999;9(1):133-146.
pubmed doi - Raha S, Robinson BH. Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem Sci. 2000;25(10):502-508.
pubmed doi - Lenaz G. The mitochondrial production of reactive oxygen species: mechanisms and implications in human pathology. IUBMB Life. 2001;52(3-5):159-164.
pubmed doi - Kim JS, He L, Lemasters JJ. Mitochondrial permeability transition: a common pathway to necrosis and apoptosis. Biochem Biophys Res Commun. 2003;304(3):463-470.
pubmed doi - Muller WE, Eckert A, Kurz C, Eckert GP, Leuner K. Mitochondrial dysfunction: common final pathway in brain aging and Alzheimer's disease—therapeutic aspects. Mol Neurobiol. 2010;41(2-3):159-171.
pubmed - Reddy PH, Manczak M, Mao P, Calkins MJ, Reddy AP, Shirendeb U. Amyloid-beta and mitochondria in aging and Alzheimer's disease: implications for synaptic damage and cognitive decline. J Alzheimers Dis. 2010;20 Suppl 2:S499-512.
pubmed - Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353-356.
pubmed doi - Hansson Petersen CA, Alikhani N, Behbahani H, Wiehager B, Pavlov PF, Alafuzoff I, Leinonen V, et al. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc Natl Acad Sci U S A. 2008;105(35):13145-13150.
pubmed doi - Parks JK, Smith TS, Trimmer PA, Bennett JP, Jr., Parker WD, Jr. Neurotoxic Abeta peptides increase oxidative stress in vivo through NMDA-receptor and nitric-oxide-synthase mechanisms, and inhibit complex IV activity and induce a mitochondrial permeability transition in vitro. J Neurochem. 2001;76(4):1050-1056.
pubmed doi - Rosales-Corral SA, Acuna-Castroviejo D, Coto-Montes A, Boga JA, Manchester LC, Fuentes-Broto L, Korkmaz A, et al. Alzheimer's disease: pathological mechanisms and the beneficial role of melatonin. J Pineal Res. 2012;52(2):167-202.
pubmed doi - Mattson MP, Liu D. Energetics and oxidative stress in synaptic plasticity and neurodegenerative disorders. Neuromolecular Med. 2002;2(2):215-231.
pubmed doi - Connern CP, Halestrap AP. Recruitment of mitochondrial cyclophilin to the mitochondrial inner membrane under conditions of oxidative stress that enhance the opening of a calcium-sensitive non-specific channel. Biochem J. 1994;302 (Pt 2):321-324.
pubmed - Reiter RJ, Tan DX. Role of CSF in the transport of melatonin. J Pineal Res. 2002;33(1):61.
pubmed doi - Facciola G, Hidestrand M, von Bahr C, Tybring G. Cytochrome P450 isoforms involved in melatonin metabolism in human liver microsomes. Eur J Clin Pharmacol. 2001;56(12):881-888.
pubmed doi - Hirata F, Hayaishi O, Tokuyama T, Seno S. In vitro and in vivo formation of two new metabolites of melatonin. J Biol Chem. 1974;249(4):1311-1313.
pubmed - Reppert SM, Weaver DR, Ebisawa T. Cloning and characterization of a mammalian melatonin receptor that mediates reproductive and circadian responses. Neuron. 1994;13(5):1177-1185.
pubmed doi - Benitez-King G. Melatonin as a cytoskeletal modulator: implications for cell physiology and disease. J Pineal Res. 2006;40(1):1-9.
pubmed doi - Urata Y, Honma S, Goto S, Todoroki S, Iida T, Cho S, Honma K, et al. Melatonin induces gamma-glutamylcysteine synthetase mediated by activator protein-1 in human vascular endothelial cells. Free Radic Biol Med. 1999;27(7-8):838-847.
pubmed doi - Reiter RJ. Oxidative damage in the central nervous system: protection by melatonin. Prog Neurobiol. 1998;56(3):359-384.
pubmed doi - Bozner P, Grishko V, LeDoux SP, Wilson GL, Chyan YC, Pappolla MA. The amyloid beta protein induces oxidative damage of mitochondrial DNA. J Neuropathol Exp Neurol. 1997;56(12):1356-1362.
pubmed doi - Pappolla MA, Sos M, Omar RA, Bick RJ, Hickson-Bick DL, Reiter RJ, Efthimiopoulos S, et al. Melatonin prevents death of neuroblastoma cells exposed to the Alzheimer amyloid peptide. J Neurosci. 1997;17(5):1683-1690.
pubmed - Malchiodi-Albedi F, Domenici MR, Paradisi S, Bernardo A, Ajmone-Cat MA, Minghetti L. Astrocytes contribute to neuronal impairment in beta A toxicity increasing apoptosis in rat hippocampal neurons. Glia. 2001;34(1):68-72.
pubmed doi - Feng Z, Zhang JT. Protective effect of melatonin on beta-amyloid-induced apoptosis in rat astroglioma C6 cells and its mechanism. Free Radic Biol Med. 2004;37(11):1790-1801.
pubmed doi - Wang J, Tung YC, Wang Y, Li XT, Iqbal K, Grundke-Iqbal I. Hyperphosphorylation and accumulation of neurofilament proteins in Alzheimer disease brain and in okadaic acid-treated SY5Y cells. FEBS Lett. 2001;507(1):81-87.
pubmed doi - Montilla-Lopez P, Munoz-Agueda MC, Feijoo Lopez M, Munoz-Castaneda JR, Bujalance-Arenas I, Tunez-Finana I. Comparison of melatonin versus vitamin C on oxidative stress and antioxidant enzyme activity in Alzheimer's disease induced by okadaic acid in neuroblastoma cells. Eur J Pharmacol. 2002;451(3):237-243.
pubmed - Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer's disease. J Neurosci. 2001;21(2):372-381.
pubmed - Matsubara E, Bryant-Thomas T, Pacheco Quinto J, Henry TL, Poeggeler B, Herbert D, Cruz-Sanchez F, et al. Melatonin increases survival and inhibits oxidative and amyloid pathology in a transgenic model of Alzheimer's disease. J Neurochem. 2003;85(5):1101-1108.
pubmed doi - Cheng X, van Breemen RB. Mass spectrometry-based screening for inhibitors of beta-amyloid protein aggregation. Anal Chem. 2005;77(21):7012-7015.
pubmed doi - Poeggeler B, Pappolla MA, Hardeland R, Rassoulpour A, Hodgkins PS, Guidetti P, Schwarcz R. Indole-3-propionate: a potent hydroxyl radical scavenger in rat brain. Brain Res. 1999;815(2):382-388.
pubmed doi - Matsubara E, Sekijima Y, Tokuda T, Urakami K, Amari M, Shizuka-Ikeda M, Tomidokoro Y, et al. Soluble Abeta homeostasis in AD and DS: impairment of anti-amyloidogenic protection by lipoproteins. Neurobiol Aging. 2004;25(7):833-841.
pubmed doi - Li XC, Wang ZF, Zhang JX, Wang Q, Wang JZ. Effect of melatonin on calyculin A-induced tau hyperphosphorylation. Eur J Pharmacol. 2005;510(1-2):25-30.
pubmed doi - Liu SJ, Wang JZ. Alzheimer-like tau phosphorylation induced by wortmannin in vivo and its attenuation by melatonin. Acta Pharmacol Sin. 2002;23(2):183-187.
pubmed - Reyes-Toso CF, Ricci CR, de Mignone IR, Reyes P, Linares LM, Albornoz LE, Cardinali DP, et al. In vitro effect of melatonin on oxygen consumption in liver mitochondria of rats. Neuro Endocrinol Lett. 2003;24(5):341-344.
pubmed - Barja G, Herrero A. Localization at complex I and mechanism of the higher free radical production of brain nonsynaptic mitochondria in the short-lived rat than in the longevous pigeon. J Bioenerg Biomembr. 1998;30(3):235-243.
pubmed doi - Genova ML, Pich MM, Bernacchia A, Bianchi C, Biondi A, Bovina C, Falasca AI, et al. The mitochondrial production of reactive oxygen species in relation to aging and pathology. Ann N Y Acad Sci. 2004;1011:86-100.
pubmed - Zhao Y, Wang ZB, Xu JX. Effect of cytochrome c on the generation and elimination of O2*- and H2O2 in mitochondria. J Biol Chem. 2003;278(4):2356-2360.
pubmed doi - Fridovich I. Superoxide radical and superoxide dismutases. Annu Rev Biochem.1995;64:97-112.
pubmed - Menendez-Pelaez A, Poeggeler B, Reiter RJ, Barlow-Walden L, Pablos MI, Tan DX. Nuclear localization of melatonin in different mammalian tissues: immunocytochemical and radioimmunoassay evidence. J Cell Biochem. 1993;53(4):373-382.
pubmed doi - Martin M, Macias M, Leon J, Escames G, Khaldy H, Acuna-Castroviejo D. Melatonin increases the activity of the oxidative phosphorylation enzymes and the production of ATP in rat brain and liver mitochondria. Int J Biochem Cell Biol. 2002;34(4):348-357.
pubmed doi - Okatani Y, Wakatsuki A, Reiter RJ, Miyahara Y. Acutely administered melatonin restores hepatic mitochondrial physiology in old mice. Int J Biochem Cell Biol. 2003;35(3):367-375.
pubmed doi - Jou MJ, Peng TI, Reiter RJ, Jou SB, Wu HY, Wen ST. Visualization of the antioxidative effects of melatonin at the mitochondrial level during oxidative stress-induced apoptosis of rat brain astrocytes. J Pineal Res. 2004;37(1):55-70.
pubmed doi - Jang MH, Jung SB, Lee MH, Kim CJ, Oh YT, Kang I, Kim J, et al. Melatonin attenuates amyloid beta25-35-induced apoptosis in mouse microglial BV2 cells. Neurosci Lett. 2005;380(1-2):26-31.
pubmed doi - Aliev G, Palacios HH, Walrafen B, Lipsitt AE, Obrenovich ME, Morales L. Brain mitochondria as a primary target in the development of treatment strategies for Alzheimer disease. Int J Biochem Cell Biol. 2009;41(10):1989-2004.
pubmed doi - Louneva N, Cohen JW, Han LY, Talbot K, Wilson RS, Bennett DA, Trojanowski JQ, et al. Caspase-3 is enriched in postsynaptic densities and increased in Alzheimer's disease. Am J Pathol. 2008;173(5):1488-1495.
pubmed doi - Garcia T, Esparza JL, Nogues MR, Romeu M, Domingo JL, Gomez M. Oxidative stress status and RNA expression in hippocampus of an animal model of Alzheimer's disease after chronic exposure to aluminum. Hippocampus. 2010;20(1):218-225.
pubmed - Danysz W, Parsons CG. The NMDA receptor antagonist memantine as a symptomatological and neuroprotective treatment for Alzheimer's disease: preclinical evidence. Int J Geriatr Psychiatry. 2003;18(Suppl 1):S23-32.
pubmed doi - Lau A, Tymianski M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. 2010;460(2):525-542.
pubmed doi - Slanar O, Pelisek V, Vanecek J. Melatonin inhibits pituitary adenylyl cyclase-activating polypeptide-induced increase of cyclic AMP accumulation and [Ca2+]i in cultured cells of neonatal rat pituitary. Neurochem Int. 2000;36(3):213-219.
pubmed doi - Brunner P, Sozer-Topcular N, Jockers R, Ravid R, Angeloni D, Fraschini F, Eckert A, et al. Pineal and cortical melatonin receptors MT1 and MT2 are decreased in Alzheimer's disease. Eur J Histochem. 2006;50(4):311-316.
pubmed - Harper DG, Stopa EG, McKee AC, Satlin A, Harlan PC, Goldstein R, Volicer L. Differential circadian rhythm disturbances in men with Alzheimer disease and frontotemporal degeneration. Arch Gen Psychiatry. 2001;58(4):353-360.
pubmed doi - Giubilei F, Patacchioli FR, Antonini G, Sepe Monti M, Tisei P, Bastianello S, Monnazzi P, et al. Altered circadian cortisol secretion in Alzheimer's disease: clinical and neuroradiological aspects. J Neurosci Res. 2001;66(2):262-265.
pubmed doi - Mishima K, Tozawa T, Satoh K, Matsumoto Y, Hishikawa Y, Okawa M. Melatonin secretion rhythm disorders in patients with senile dementia of Alzheimer's type with disturbed sleep-waking. Biol Psychiatry. 1999;45(4):417-421.
pubmed doi - Van Someren EJ. Circadian rhythms and sleep in human aging. Chronobiol Int. 2000;17(3):233-243.
pubmed doi - Cohen-Mansfield J, Garfinkel D, Lipson S. Melatonin for treatment of sundowning in elderly persons with dementia - a preliminary study. Arch Gerontol Geriatr. 2000;31(1):65-76.
pubmed doi - Yamadera H, Ito T, Suzuki H, Asayama K, Ito R, Endo S. Effects of bright light on cognitive and sleep-wake (circadian) rhythm disturbances in Alzheimer-type dementia. Psychiatry Clin Neurosci. 2000;54(3):352-353.
pubmed doi - Uchida K, Okamoto N, Ohara K, Morita Y. Daily rhythm of serum melatonin in patients with dementia of the degenerate type. Brain Res. 1996;717(1-2):154-159.
pubmed doi - Ohashi Y, Okamoto N, Uchida K, Iyo M, Mori N, Morita Y. Daily rhythm of serum melatonin levels and effect of light exposure in patients with dementia of the Alzheimer's type. Biol Psychiatry. 1999;45(12):1646-1652.
pubmed doi - Wu YH, Feenstra MG, Zhou JN, Liu RY, Torano JS, Van Kan HJ, Fischer DF, et al. Molecular changes underlying reduced pineal melatonin levels in Alzheimer disease: alterations in preclinical and clinical stages. J Clin Endocrinol Metab. 2003;88(12):5898-5906.
pubmed doi - Zhou JN, Liu RY, Kamphorst W, Hofman MA, Swaab DF. Early neuropathological Alzheimer's changes in aged individuals are accompanied by decreased cerebrospinal fluid melatonin levels. J Pineal Res. 2003;35(2):125-130.
pubmed doi - Skene DJ, Swaab DF. Melatonin rhythmicity: effect of age and Alzheimer's disease. Exp Gerontol. 2003;38(1-2):199-206.
pubmed doi - Magri F, Locatelli M, Balza G, Molla G, Cuzzoni G, Fioravanti M, Solerte SB, et al. Changes in endocrine circadian rhythms as markers of physiological and pathological brain aging. Chronobiol Int. 1997;14(4):385-396.
pubmed doi - Maurizi CP. Choroid plexus portals and a deficiency of melatonin can explain the neuropathology of Alzheimer's disease. Med Hypotheses. 2010;74(6):1059-1066.
pubmed doi - McCurry SM, Reynolds CF, Ancoli-Israel S, Teri L, Vitiello MV. Treatment of sleep disturbance in Alzheimer's disease. Sleep Med Rev. 2000;4(6):603-628.
pubmed doi - Brusco LI, Marquez M, Cardinali DP. Melatonin treatment stabilizes chronobiologic and cognitive symptoms in Alzheimer's disease. Neuro Endocrinol Lett. 2000;21(1):39-42.
pubmed - Brusco LI, Marquez M, Cardinali DP. Monozygotic twins with Alzheimer's disease treated with melatonin: Case report. J Pineal Res. 1998;25(4):260-263.
pubmed doi - Mishima K, Okawa M, Hozumi S, Hishikawa Y. Supplementary administration of artificial bright light and melatonin as potent treatment for disorganized circadian rest-activity and dysfunctional autonomic and neuroendocrine systems in institutionalized demented elderly persons. Chronobiol Int. 2000;17(3):419-432.
pubmed doi - Mahlberg R, Kunz D, Sutej I, Kuhl KP, Hellweg R. Melatonin treatment of day-night rhythm disturbances and sundowning in Alzheimer disease: an open-label pilot study using actigraphy. J Clin Psychopharmacol. 2004;24(4):456-459.
pubmed doi - Asayama K, Yamadera H, Ito T, Suzuki H, Kudo Y, Endo S. Double blind study of melatonin effects on the sleep-wake rhythm, cognitive and non-cognitive functions in Alzheimer type dementia. J Nihon Med Sch. 2003;70(4):334-341.
pubmed doi - Singer C, Tractenberg RE, Kaye J, Schafer K, Gamst A, Grundman M, Thomas R, et al. A multicenter, placebo-controlled trial of melatonin for sleep disturbance in Alzheimer's disease. Sleep. 2003;26(7):893-901.
pubmed - Gehrman PR, Connor DJ, Martin JL, Shochat T, Corey-Bloom J, Ancoli-Israel S. Melatonin fails to improve sleep or agitation in double-blind randomized placebo-controlled trial of institutionalized patients with Alzheimer disease. Am J Geriatr Psychiatry. 2009;17(2):166-169.
pubmed doi - Lauterbach EC, Victoroff J, Coburn KL, Shillcutt SD, Doonan SM, Mendez MF. Psychopharmacological neuroprotection in neurodegenerative disease: assessing the preclinical data. J Neuropsychiatry Clin Neurosci. 2010;22(1):8-18.
pubmed doi - Kato K, Hirai K, Nishiyama K, Uchikawa O, Fukatsu K, Ohkawa S, Kawamata Y, et al. Neurochemical properties of ramelteon (TAK-375), a selective MT1/MT2 receptor agonist. Neuropharmacology. 2005;48(2):301-310.
pubmed doi - Stevenson S,Bryson S, Amayke D et al. Study to investigate the absolute bio availabilityof a single oral dose of ramelteon(TAK-375)in healthy male subjects. Clin Pharmacol Ther 2004;75:22.
- Pandi-Perumal SR, Srinivasan V, Poeggeler B, Hardeland R, Cardinali DP. Drug Insight: the use of melatonergic agonists for the treatment of insomnia-focus on ramelteon. Nat Clin Pract Neurol. 2007;3(4):221-228.
pubmed doi - Pandi-Perumal SR, Srinivasan V, Spence DW, Moscovitch A, Hardeland R, Brown GM, Cardinali DP. Ramelteon: a review of its therapeutic potential in sleep disorders. Adv Ther. 2009;26(6):613-626.
pubmed doi - Pandi-Perumal SR, Spence DW,. Verster JC, Srinivasan V, Brown GM , Cardinali DP, Hardeland R. Pharmacotherapy of insomnia with ramelteon.: safety, efficacy and clinical applications. J Central Nerv Dis 2011;3:51-65.
- Lauterbach EC, Shillcutt SD, Victoroff J, Coburn KL, Mendez MF. Psychopharmacological neuroprotection in neurodegenerative disease: heuristic clinical applications. J Neuropsychiatry Clin Neurosci. 2010;22(2):130-154.
pubmed doi - Imbesi M, Uz T, Dzitoyeva S, Manev H. Stimulatory effects of a melatonin receptor agonist, ramelteon, on BDNF in mouse cerebellar granule cells. Neurosci Lett. 2008;439(1):34-36.
pubmed doi
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Neurology Research is published by Elmer Press Inc.