

| Journal of Neurology Research, ISSN 1923-2845 print, 1923-2853 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Neurol Res and Elmer Press Inc |
| Journal website https://www.neurores.org |
Review
Volume 13, Number 1, September 2023, pages 1-11

High Wall Shear Incites Cerebral Aneurysm Formation and Low Wall Shear Stress Propagates Cerebral Aneurysm Growth
Vivig Shantha Kumara, b, Vignarth Shantha Kumara
aInternal Medicine, California Institute of Behavioral Neurosciences and Psychology, Fairfield, CA, USA
bCorresponding Author: Vivig Shantha Kumar, Internal Medicine, California Institute of Behavioral Neurosciences and Psychology, Fairfield, CA 94534, USA
Manuscript submitted January 30, 2023, accepted March 8, 2023, published online August 8, 2023
Short title: Cerebral Hypertension and Hemodynamic Patterns
doi: https://doi.org/10.14740/jnr749
- Abstract
- Introduction
- Vasculoprotective Effect of Normal Laminar Shear Stress
- Vascular Endothelial Cell Structural Changes in Response to High Shear Stress
- Activation of Endothelial Cell Proinflammatory Response by High WSS
- Growing and Thin End of CAs Demonstrate Low WSS
- Low WSS Enhances Inflammatory Cell Accumulation and Endothelial Cell Loss Contributing to Vessel Wall Weakening and Rupture
- Conclusions
- References
| Abstract | ▴Top |
This review discusses mechanisms for the development of cerebral aneurysms. Endothelial cells exhibit a variety of structural and functional changes when they come into contact with normal laminar flow. In response to laminar shear stress, endothelial cells modify their potassium ion channels, go through cytoskeletal rearrangements and shape modifications and create prostacyclin. In cerebral arteries, aneurysmal dilatation most frequently starts at locations with substantial wall shear stress, which include arterial bifurcations and vascular branch sites, where blood flow abruptly switches to turbulent flow. At this point, high shear stress frequently arises, placing increased strain on the vasculature. As the vascular branch points and arterial bifurcations are the initial sites of cerebral aneurysm genesis, this helps confirm the role of high wall shear stress in the development of cerebral aneurysms. Low wall shear stress increases the initial proinflammatory effect already present in the vasculature, which furthers the formation of cerebral aneurysms. In fact, regions of aneurysmal regions with low wall shear stress grow more quickly and are more prone to rupture compared to regions with high wall shear stress. Therefore, it seems plausible to assume that turbulent blood flow inside a dilated cerebral aneurysm causes low wall shear stress, thereby encouraging aneurysmal growth.
Keywords: Cerebral aneurysm; Hemodynamic disturbances; Wall shear stress; Aneurysmal growth; Aneurysmal rupture
| Introduction | ▴Top |
A confined, outward pathological dilatation of the artery wall, cerebral aneurysm (CA) is thought to affect 1-3% of people in the general population [1]. CA development and growth follows an amalgamation of multiple insults with individual contributions from hemodynamic stress and inflammatory pathways [2, 3]. Substantial clinical and experimental experiences demonstrate a role of hemodynamic influences in CA pathogenesis, suggesting that altered hemodynamics modulate a biphasic response defined by early initial CA formation and later cerebral growth [4-8]. A series of actions connected to stages of arterial wall remodeling in response to hemodynamic stresses are represented by the initiation, growth, and rupture of CAs [9, 10]. Hemodynamic-induced endothelial dysfunction is a starting point for the development of CAs. Varying patterns of blood flow exert mechanical stresses on vascular endothelial cells, altering the functions of these cells and predisposing to vessel wall changes [11-14]. The primary hemodynamic cause of CA growth, formation, and rupture is wall shear stress (WSS). High shear stress in arterial branch points and bifurcations coincides with histological markers of nascent CA formation [15, 16]. Cerebral vessels at these locations commonly demonstrate early destructive vessel wall changes such as, most commonly, damage to endothelial cells with signs of either altered protective endothelial cell phenotype or endothelial cell loss and fragmentation of the internal elastic lamina (IEL). Low WSS, on the other hand, is commonly observed at the growing end of CAs such as the neck and dome regions of CAs [17, 18]. These regions are observed to demonstrate marked inflammatory vessel wall remodeling demonstrated by increased macrophage tissue trafficking, release of macrophage derived products (matrix metalloproteinases (MMPs)), and loss of smooth muscle cells (SMCs). Therefore, our main objective in this study is to understand the contributions of varying hemodynamic perturbations toward the development and growth of CAs.
| Vasculoprotective Effect of Normal Laminar Shear Stress | ▴Top |
Under physiological states, cerebral blood vessels display a laminar flow pattern. Laminar flow refers to a unidirectional, orderly pattern characterized by parallel vectors. In order to control a number of vascular processes, endothelial cells’ reactions to normal fluid shear stress are crucial. Endothelial cells that are subjected to a typical laminar flow and typical WSS show a number of structural and functional modifications. Under normal physiological laminar shear stress, endothelial cells adopt an anti-inflammatory and nonproliferative surface expression characterized by increased resistance to inflammation, growth, and apoptosis [19]. Laminar shear stress modulates vascular tone through its influence on the production of nitric oxide (NO) [20, 21]. Endothelial nitric oxide synthase (eNOS) modulates local production of NO and is activated through phosphorylation of protein kinase B in response to laminar stress, leading to upregulation of eNOS activity [22, 23]. Besides upregulating eNOS activity through protein kinase B phosphorylation, laminar WSS induces continuous eNOS mRNA transcription through the c-Src-dependent pathways [24]. Additionally, in endothelial cell cultures, laminar shear stress induces Kruppel-like factor 2 (KLF2), which contributes to NO-dependent vasodilation [25, 26]. Endothelial cell overexpression of KLF2 abundantly induces eNOS expression [27, 28]. Laminar fluid shear stress mediates an antithrombotic and anti-inflammatory effect through the upregulation of KLF2 [29, 30]. Induced by laminar shear stress, KLF2 reduced the expression of the pro-inflammatory adhesion molecules vascular cell adhesion molecule (VCAM)-1 and E-selectin in endothelial cells [31]. Further, endothelial cells introduced with KLF2 were found to display decreased attachment of white blood cells in vitro flow assay studies [31]. Likewise, expression of the nuclear factor kappa B (NF-κB) ligand is downregulated [32, 33]. Decreased expression of NF-κB, a proinflammatory transcription factor, minimizes the development of a proinflammatory extracellular environment within the vascular wall. Also, laminar shear stress mediates antithrombotic responses through KLF2. Endothelial cells in normal vessels adapt an anticoagulant response to laminar shear stress by upregulating thrombomodulin, heparin sulfate, and tissue factor inhibitor [34]. The expression of thrombomodulin is also continuously increased by laminar shear stress, but it increases by a factor of two more than it does in normal cells [35]. Furthermore, shear stress increases endothelial expression of tissue plasminogen activators while suppressing plasminogen activator inhibitor type 1 release [36].
Laminar shear stress also promotes cell cycle arrest in the G1 or G0 phase, which keeps endothelial cells in a quiescent condition [33]. Endothelial cell intracellular processes such as gene transcription, protein synthesis, cell proliferation, and ultimately cytoskeletal rearrangement and morphological changes are also regulated by normal physiological shear stress [37, 38]. The mitogen-activated protein kinase (MAPK) family of proteins is one of the most significant signaling pathways mediating endothelial cell proliferative response to laminar WSS [39, 40]. MAPK proteins (ERK ½, p38 and JNK) activated in response to shear stress facilitate conduct of extracellular signals into the cell nucleus, where they influence gene transcription [22, 41, 42]. The net effect of MAPK activation is the ultimate activation of ERK ½ leading to protein synthesis, cell proliferation and an inhibition of apoptosis [43-45]. Additionally, cyclin dependent-kinase, responsible for vascular endothelial cell proliferation, is suppressed [22]. Repression of endothelial cyclin dependent kinase prevents aberrant cell proliferation resulting in a healthy balance between proliferation between maintenance. Moreover, the antimitotic pathway of 5’-AMP-activated protein kinase (AMPK) and the proliferative pathway of AKT is simultaneously activated. Dual activation of both the AMPK/AKT pathway maintains a balanced expression of mammalian target of rapamycin (mTOR) signaling, a molecular pathway governing vascular endothelial cell proliferation [46]. Ultimately, through these molecular signaling pathways, endothelial cells remain in a quiescent antiproliferative state secondary to an arrest of the cell cycle in either the G1 or G0 phase promoting indefinite endothelial cell survival.
| Vascular Endothelial Cell Structural Changes in Response to High Shear Stress | ▴Top |
The frequent occurrence of CAs in vascular branch points and bifurcation points emphasizes the significance of hemodynamic stresses in the beginning of CA formation [47-51]. Indeed, there is a higher prevalence of CAs in association with morphological abnormalities of the cerebral vasculature, such as hypoplasia/occlusion of a section of the circle of Willis or arteriovenous malformations that provide elevated flow patterns and high WSS locally [47, 52-55]. Aneurysmal dilation of cerebral vessels most commonly begins at sites of high WSS. High WSS commonly develops at arterial vascular branch points and arterial bifurcations, where blood flow suddenly changes from the steady uniform laminar pattern into a more chaotic turbulent pattern exerting greater tension on the vascular wall. Several pieces of animal studies highlight a central role of altered hemodynamics in the initiation of CA formation. Elevation of the WSS beyond threshold conditions, from observations in several animal models, documents histopathological vascular wall changes suggestive of early CA formation: fragmentation of the IEL and endothelial cell phenotype modulation [48, 56]. From histopathological examination of affected cerebral blood vessels, Steiger et al deduced that experimentally induced sustained elevations of WSS is attended by a fragmentation of the IEL of blood vessels [56]. Similarly, Stehbens et al noted that, in addition, endothelial cells show an alteration in their normal phenotype as well as endothelial damage [48]. Gao et al using a rabbit model, demonstrated a drastic nine-fold increase in basilar artery flow following ligation of the common carotid artery. Additionally, newly formed CAs were noted at the basilar artery bifurcation, characterized histologically by a loss of the internal elastic media and an outward bulged and thinned tunica media [57]. Dogs’ carotid arteries were ligated experimentally to create new branch points, and Meng et al observed remodulative changes at these bifurcations that resembled the beginning of an intracranial aneurysm, including disruption of the IEL, loss of medial SMCs, and a decreased proliferation of SMCs [58]. Further, Jamous et al studied CA occurring at high flow bifurcation sites and documented endothelial cell morphological alterations during the early phase of aneurysm development. In the early phase of CA development, endothelial cells were observed to have an abnormal endothelial cell morphology ranging from segmental detachment of the endothelial cell plasma membrane to endothelial cell deformation with a vacuolated cytoplasm and/or nucleus depending on the degree of destructive remodeling [59]. Fukuda et al [60] similarly observed that high wall shear incites CA formation and endothelial cell injury at sites of nascent CA formation, similarly, corresponding to endothelial cell structural modifications as described by Jamous et al [59]. One such study performed by Fukuda et al correlated aneurysmal degenerative changes in endothelial cells with the magnitude of WSS in variable areas of the cerebral blood vessel. Herein, it was discovered that in the region of the vessel bifurcation, the intimal endothelial cells showed characteristic initial changes suggestive of early progression to aneurysm dilation. Given the bifurcation of the vessels at this site, it was noted that the intima of these vessels experienced the highest magnitude of WSS [60]. Along the same lines, observations from animal studies still further strengthen the positive correlation between a high WSS and early aneurysmal changes. Moreover, in experimental models of CA formation in rats and primates, increased cerebral blood flow and hypertension were necessary prerequisites for aneurysmal dilation [50, 61-63]. In general, these studies conclude that aneurysm initiation starts with deranged initial endothelial cell responses leading to structural and functional modifications of the endothelium.
| Activation of Endothelial Cell Proinflammatory Response by High WSS | ▴Top |
Flow acceleration at bifurcation points produces a hemodynamic environment characterized by high WSS which triggers initiation of aneurysmal dilation. In experimental models of CAs [4, 15-17], increased cerebral blood flow and systemic hypertension are necessary prerequisites for the initiation of CA formation (Table 1) [48, 56, 58, 60, 64-73]. Likewise, Kulcsar et al analyzed the hemodynamics of a cerebral vasculature in three patients before and after the development of an intracranial aneurysm and observed that intracranial aneurysm consistently formed at locations characterized by high WSS [74]. Further, Metaxa et al using rabbit models noted the occurrence of nascent aneurysm formation at the basilar terminus region following basilar artery flow increase, a region of elevated WSS [75]. High WSS is exceedingly implicated in destructive vessel remodeling and endothelial cells at arterial bifurcations, suggested by the fact that vascular branch points and apices become progressively dysfunctional following prolonged abnormal hemodynamic stresses [19]. Increased wall shear and excessive hemodynamic stresses activate endothelial cell mechanoreceptors leading to increased signal transduction and activation of inflammatory pathways, leading to destructive inflammatory vessel wall remodeling. High WSS, however, evokes a proinflammatory, procoagulant and proliferative phenotype predisposing to vascular remodeling. Activation of NF-κB, a proinflammatory transcription factor, in endothelial cells challenged with hemodynamic stress activates inflammatory signaling pathways leading to CA initiation [76] NF-κB, a proinflammatory transcription factor, is activated by increased shear stress on endothelial cells, regulating the expression of various proinflammatory genes [77-81]. In vivo experimental models of CAs observed that increased flow and hypertension are necessary prerequisites for NF-κB activation in rat models of aortic aneurysms [82]. In response to turbulent flow, NF-κB activation occurs predominantly in the endothelial cells and macrophages. In quiescent unstimulated cells, NF-κB is tucked away in the cytoplasm in combination with inhibitor IKB proteins, preventing its translocation into the nucleus. Appropriate activating signals phosphorylate IKB, acting to abborate the inhibitory anti-migratory influence of IKB. NF-κB subsequently translocates to the nucleus to evoke the transcription of proinflammatory genes [76]. Cultured endothelial cells demonstrate increased nuclear translocation of NF-κB in response to fluid shear stress by activating IKB kinase through phosphorylation [83]. Further, use of antibody directed against the p65 nuclear localization signal subunit of NF-κB demonstrated increased localization and activation of NF-κB in the arterial walls with the use of immunohistochemical studies [84]. Likewise, use of immunostaining and western-blot analysis techniques confirmed that NF-κB was significantly phosphorylated and activated in the vascular endothelial cells and macrophages during the initiation of CAs in murine rat models of CA [85-87]. Animal models clearly advocate that NF-κB activation at the site of vascular injury is necessary for the formation of intracranial aneurysms. Mice devoid of NF-κB expression were observed to have a significant blockade of aneurysm formation. NF-κB plays a critical developmental role in the genesis of nascent CAs by regulating the transcription of downstream pro-inflammatory genes leading to phenotypic alterations of the vascular endothelium [64, 88]. The major downstream target of NF-κB following activation is the upregulation of proinflammatory adhesion molecules, VCAM-1 and monocyte chemotactic protein (MCP)-1, on the vascular endothelium leading to increased neutrophil and macrophage tissue trafficking. Macrophage infiltration results in the release of MMPs (MMP-2 and MMP-9), capable of proteolytically degrading the extracellular matrix, as well as the induction of inducible nitric oxide synthase (iNOS) leading to the pathological formation of NO mediating vascular SMC apoptosis.
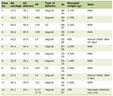 Click to view | Table 1. High Wall Shear Stress (WSS): Summary of Hemodynamic Pattern, Growth Rate, and Vascular Wall Changes |
| Growing and Thin End of CAs Demonstrate Low WSS | ▴Top |
Evidence implicating low WSS in the growth of CA development is suggested by several studies. Cebral et al [89] initially inferred that local propagation and growth of CAs is driven by interactions between regional blood flow and the vascular wall. Several experimental in vitro studies, subsequently, collectively agree on low blood flow velocity as a critical driving force [17, 89, 90]. Watton et al observed continuous enlargement of the growing end of CAs due to a low WSS following an initial aneurysmal bulge enlargement [91]. Tateshima et al using middle CAs with the development of an enlarged bleb measured local WSS patterns and observed that the enlarged bleb end of a growing aneurysm displayed low WSS [92]. Likewise, Kadasi et al, using computational fluid dynamic models of 16 CAs identified during surgery noted that the occurrence of low WSS coincided with the thinner growing regions of the aneurysmal wall [93]. Shojma et al in his computational analysis of 20 CAs affecting human middle cerebral arteries consistently observed that low WSS was noted at the apex of the CA, a region of the aneurysm corresponding to the height of aneurysmal growth [4]. Similar conclusions regarding the relationship between WSS and aneurysmal growth was explored by Boussel et al, who suggested that regions of the CA with low WSS experienced higher rates of aneurysmal growth compared to regions of the CA with high WSS, which experienced lower rates of aneurysmal growth [94]. Further observations highlighting the contribution of low WSS in the propagation and increase in size of CAs comes from the study of Skodvin et al [95], which studied the relationship between low WSS and the risk of rupture. Here, it was observed that CAs that displayed larger areas of low WSS were more likely to rupture, indirectly suggesting a positive correlation between low wall stress and aneurysmal size [95].
Further examination of the molecular cross-talk between low blood flow velocity and CA growth is linked to structural changes of vascular endothelial cells. Endothelial cells, exposed to patterns of low WSS, are characterized by decreased cell-cell adhesion, endothelial cell loss, and thrombus formation, which are necessary prerequisites for the structural weakening of the aneurysmal wall [94, 96, 97]. After initiation of CA formation, the region of blood vessels exposed to high WSS demonstrates fragmentation and loss of the IEL mediated by MMPs [56, 58]. Given the fact that the IEL contributes significantly to the structural integrity of the vessel wall, destruction of the IEL leads to an initial outward bulge creating local flow patterns of stagnant flow and low WSS. Aneurysmal bulge development exposes the growing end of the aneurysmal sac to low WSS, accelerating the previously initiated proinflammatory response by the vascular endothelium in response to high WSS. The predominance of low WSS in high growth regions of CAs defines a dominant function of low WSS in the continued growth and expansion of CAs; however, studies highlighting plausible molecular mechanisms governing this growth are relatively sparse [4, 91-94]. Endothelial cells exposed to sustained periods of low WSS respond by increasing proliferation of endothelial cells, triggering apoptosis of endothelial cells, upregulating proinflammatory and procoagulant mediators, increasing production of vasoconstrictive agents and decreasing production of vasodilatory mediators and antioxidative agents [98]. The ensuing endothelial dysfunction triggers the upregulation of adhesion molecules (VCAM-1 and intercellular adhesion molecule (ICAM)-1) and proinflammatory cytokines (tissue necrosis factor (TNF)-α, interleukin (IL)-1) and reactive oxygen species on the luminal cell surface [94, 99]. Further, low WSS increases endothelial expression of NF-κB ligand, a proinflammatory transcription factor. Increased transcription of the NF-κB pathway provides for increased adhesion and infiltration of leukocytes to the vascular endothelium. Leukocyte trafficking into the arterial wall allows for the release of proteases and proinflammatory cytokines that degrade the structural matrix and induce vascular smooth muscle apoptosis [100]. The weakened media in the arterial wall subsequently facilitates aneurysmal dilation under low WSS. Additionally, low WSS reduces mechanical stimulation and deformation of the vascular endothelium, resulting in an impaired synthesis and secretion of NO from the endothelium. Decreased expression of NO on the intimal surface promotes vasoconstriction and platelet aggregation [101, 102]. Consequently, increased inflammatory cell adhesion as well as aggregation of red blood cells and platelets damages the intima, resulting in intimal inflammation [103, 104]. As such, following injury inflammation of the intima, upregulation of inflammatory cell adhesion proteins is subsequently attended by increased leukocyte trafficking into the vascular wall. In contrast to a high WSS environment not favorable for tissue trafficking of leukocytes due to insufficient residence time in the vasculature, a low wall shear facilitates leukocyte transmigration due to the presence of a pro-adhesive endothelium in conjunction with increased residence time in the vasculature [105]. The resulting inflammatory cell infiltrates structurally degrades the extracellular matrix by releasing MMPs (MMP-2 and MMP-9). Additionally, following aneurysm initiation, the dome experiences low levels of WSS as a result of regional blood flow stagnation. Local stagnation of blood flow prevents shear stress-induced eNOS action leading to a dysfunction of flow induced NO synthesis. Decreased synthesis of endothelial derived NO triggers apoptosis of vascular SMCs setting into motion, the process of vessel wall remodeling [60, 106-108].
Collectively these studies suggest that areas of an CA displaying low WSS experience greater rates of growth and are more prone to rupture compared to areas of an aneurysm that display high WSS. So, it is safe to assume that low WSS generated by turbulent blood flow within a dilated CA functions to propagate aneurysmal growth.
| Low WSS Enhances Inflammatory Cell Accumulation and Endothelial Cell Loss Contributing to Vessel Wall Weakening and Rupture | ▴Top |
Low levels of WSS evoke a proinflammatory endothelial cell phenotype leading to aneurysmal growth, progression, and rupture. In response to a laminar, physiological level of shear stress, endothelial cells adopt a nonproliferative and noninflammatory phenotype. Following initiation of nascent CA formation, aneurysmal dilation creates turbulent flow patterns characterized by fewer organized parallel flow vectors, exposing the endothelium of the growing sac to lower WSS. Low wall shear inside the growing end of the aneurysmal sac evokes an atherogenic response by promoting expression of a proinflammatory endothelial cell phenotype [109-112]. Moreover, low wall shear modifies the secretory response of endothelial cells, characterized by decreased production of vasodilators (NO and prostacyclin) and antioxidants (superoxide dismutase) and increased production of vasoconstrictors (endothelin-1), reactive oxygen species and proinflammatory cytokines (TNF-α and IL-1B) [89]. Endothelial cells increase synthesis and release of reactive oxygen species and proinflammatory cytokines and upregulate proinflammatory cell surface adhesion molecules (VCAM-1, ICAM-1) on the luminal cell surface [105]. Further, low shear stress facilitates apoptosis of endothelial cells with a weakening of the aneurysmal wall. Indeed, in a comparative study between ruptured and unruptured CAs, ruptured CAs were observed to show increased rates of apoptosis [112, 113]. Moreover, areas of low WSS coincided with thin wall regions of the CA such as the dome. The net outcome achieved [58] is increased inflammatory cell infiltration, MMP production, SMC proliferation and migration leading to weakening of the vessel wall and aneurysmal rupture (Table 2) [4, 72, 92, 94-97, 114-119].
Click to view | Table 2. Low Wall Shear Stress (WSS) and Cerebral Aneurysms (CAs): Summary of Hemodynamic Pattern, Growth Rate, and Vascular Wall Changes |
Building on this, lymphocytes play a vital role in the rupture of aneurysms [11]. T lymphocytes propagate the destructive inflammatory process through the elaboration of proinflammatory cytokines (TNF and interferon (IFN)-γ) leading to activation of macrophages, B lymphocytes and upregulation of surface adhesion molecules. Sawyer et al observed that following initiation of intracranial aneurysms in experimental hypertension molecules, lymphocyte-depleted mice developed significantly fewer aneurysms compared to lymphocyte-rich mice [120]. In addition, lymphocyte release of IFN-γ, a potent inducer of macrophage activation, determines the course of aneurysmal growth and rupture [11, 121]. In a comparative study, macrophage infiltration was shown to be significantly correlated with an increased risk of rupture in ruptured CAs compared to unruptured CAs [122-124]. Further, macrophage-derived proteases (MMPs 1, 2, and 9) are consistently overexpressed in aneurysmal walls and ruptured aneurysms show a higher expression of MMP-2 and MMP-9 compared to unruptured aneurysms [123]. Similarly, Sawyer et al demonstrated that lymphocyte-depleted mice had lower levels of MMP-2 and MMP-9 compared to lymphocyte-rich mice and consequently had lower risk of CA rupture [120]. Kurki et al in a comparative study between ruptured and unruptured cerebral aneurysm models using oligonucleotide microarrays to analyze endothelial cell gene expression in varying hemodynamic stress observed increased inflammatory cell chemotaxis and leukocyte trafficking, oxidative stress, extracellular matrix degradation and destructive vascular remodeling in ruptured aneurysms as opposed to unruptured aneurysms [125]. Apart from mediating proinflammatory changes on the vessel wall, low WSS exerts detrimental vascular wall remodeling changes by influencing endothelial cell expression of NO. In response to low WSS, endothelial cells suppress expression of endothelial-derived NO [126]. Given the vital role of NO in vascular physiology such as regulation of vascular tone, inhibition of SMC proliferation, decreased production of proinflammatory mediators, loss of NO has detrimental effects of aneurysmal growth [127]. Aneurysmal wall devoid of adequate NO production displays increased oxidative stress due to an increase in oxidase activity unbalanced by appropriate superoxide scavenger activity [128].
Ultimately, in regions of the vessel wall displaying atherosclerotic and hyperplastic changes as well in aneurysmal rupture, low WSS was demonstrated, highlighting a pivotal role of low WSS in inducing a proinflammatory atherogenic response culminating in vessel wall weakening and subsequent aneurysmal rupture.
| Conclusions | ▴Top |
It has been discovered that CA regions with low WSS expand faster and are more likely to burst than those with high WSS. Consequently, it is plausible to infer that low WSS, which in turn promotes aneurysmal growth, is caused by turbulence in the blood flow within a dilated CA. The preponderance of low WSS in high growth regions of CAs suggests that low WSS plays a prominent role in the continuing growth and expansion of CAs; nevertheless, research showing probable molecular processes behind this growth are rare. Low WSS contributes to the formation and expansion of CAs, starting with its effects on vascular endothelial cells. In response to sustained low WSS, endothelial cells multiply more, cause death in endothelial cells, increase pro-inflammatory and procoagulant mediators, produce more vasoconstrictive agents, and decrease the production of antioxidative and vasodilatory mediators, culminating in a destructive cascade of vessel wall remodeling unable to tolerate hemodynamic stresses leading to aneurysmal growth and eventual rupture.
Acknowledgments
The authors declare that there are no acknowledgments to declare.
Financial Disclosure
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of Interest
The authors declare that there is no conflict of interest.
Author Contributions
Vivig Shantha Kumar was responsible for writing the discussion, incorporating the tables and references and proofreading the article.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Sheinberg DL, McCarthy DJ, Elwardany O, Bryant JP, Luther E, Chen SH, Thompson JW, et al. Endothelial dysfunction in cerebral aneurysms. Neurosurg Focus. 2019;47(1):E3.
doi - Brisman JL, Song JK, Newell DW. Cerebral aneurysms. N Engl J Med. 2006;355(9):928-939.
doi - Schievink WI. Intracranial aneurysms. N Engl J Med. 1997;336(1):28-40.
doi - Shojima M, Oshima M, Takagi K, Torii R, Hayakawa M, Katada K, Morita A, et al. Magnitude and role of wall shear stress on cerebral aneurysm: computational fluid dynamic study of 20 middle cerebral artery aneurysms. Stroke. 2004;35(11):2500-2505.
doi - Jou LD, Lee DH, Morsi H, Mawad ME. Wall shear stress on ruptured and unruptured intracranial aneurysms at the internal carotid artery. AJNR Am J Neuroradiol. 2008;29(9):1761-1767.
doi pubmed pmc - Castro M, Putman C, Radaelli A, Frangi A, Cebral J. Hemodynamics and rupture of terminal cerebral aneurysms. Acad Radiol. 2009;16(10):1201-1207.
doi - Sforza DM, Putman CM, Cebral JR. Hemodynamics of cerebral aneurysms. Annu Rev Fluid Mech. 2009;41:91-107.
doi pubmed pmc - Takao H, Murayama Y, Otsuka S, Qian Y, Mohamed A, Masuda S, Yamamoto M, et al. Hemodynamic differences between unruptured and ruptured intracranial aneurysms during observation. Stroke. 2012;43(5):1436-1439.
doi - Hoh BL, Rabinov JD, Pryor JC, Ogilvy CS. A modified technique for using elastase to create saccular aneurysms in animals that histologically and hemodynamically resemble aneurysms in human. Acta Neurochir (Wien). 2004;146(7):705-711.
doi - Tada Y, Kanematsu Y, Kanematsu M, Nuki Y, Liang EI, Wada K, Makino H, et al. A mouse model of intracranial aneurysm: technical considerations. Acta Neurochir Suppl. 2011;111:31-35.
doi pubmed pmc - Chyatte D, Bruno G, Desai S, Todor DR. Inflammation and intracranial aneurysms. Neurosurgery. 1999;45(5):1137-1146; discussion 1146-1137.
doi - Frosen J, Piippo A, Paetau A, Kangasniemi M, Niemela M, Hernesniemi J, Jaaskelainen J. Remodeling of saccular cerebral artery aneurysm wall is associated with rupture: histological analysis of 24 unruptured and 42 ruptured cases. Stroke. 2004;35(10):2287-2293.
doi - Hashimoto T, Meng H, Young WL. Intracranial aneurysms: links among inflammation, hemodynamics and vascular remodeling. Neurol Res. 2006;28(4):372-380.
doi pubmed pmc - Aoki T, Kataoka H, Ishibashi R, Nozaki K, Hashimoto N. Gene expression profile of the intima and media of experimentally induced cerebral aneurysms in rats by laser-microdissection and microarray techniques. Int J Mol Med. 2008;22(5):595-603
- Valencia A, Morales H, Rivera R, Bravo E, Galvez M. Blood flow dynamics in patient-specific cerebral aneurysm models: the relationship between wall shear stress and aneurysm area index. Med Eng Phys. 2008;30(3):329-340.
doi - Rhoton AL, Jr. Aneurysms. Neurosurgery. 2002;51(4 Suppl):S121-S158
- Tanoue T, Tateshima S, Villablanca JP, Vinuela F, Tanishita K. Wall shear stress distribution inside growing cerebral aneurysm. AJNR Am J Neuroradiol. 2011;32(9):1732-1737.
doi pubmed pmc - Kadirvel R, Ding YH, Dai D, Zakaria H, Robertson AM, Danielson MA, Lewis DA, et al. The influence of hemodynamic forces on biomarkers in the walls of elastase-induced aneurysms in rabbits. Neuroradiology. 2007;49(12):1041-1053.
doi - Soldozy S, Norat P, Elsarrag M, Chatrath A, Costello JS, Sokolowski JD, Tvrdik P, et al. The biophysical role of hemodynamics in the pathogenesis of cerebral aneurysm formation and rupture. Neurosurg Focus. 2019;47(1):E11.
doi - Marletta MA. Nitric oxide: biosynthesis and biological significance. Trends Biochem Sci. 1989;14(12):488-492.
doi - Nishida K, Harrison DG, Navas JP, Fisher AA, Dockery SP, Uematsu M, Nerem RM, et al. Molecular cloning and characterization of the constitutive bovine aortic endothelial cell nitric oxide synthase. J Clin Invest. 1992;90(5):2092-2096.
doi pubmed pmc - Li YS, Haga JH, Chien S. Molecular basis of the effects of shear stress on vascular endothelial cells. J Biomech. 2005;38(10):1949-1971.
doi - Go YM, Boo YC, Park H, Maland MC, Patel R, Pritchard KA, Jr., Fujio Y, et al. Protein kinase B/Akt activates c-Jun NH(2)-terminal kinase by increasing NO production in response to shear stress. J Appl Physiol (1985). 2001;91(4):1574-1581.
doi - Davis ME, Cai H, Drummond GR, Harrison DG. Shear stress regulates endothelial nitric oxide synthase expression through c-Src by divergent signaling pathways. Circ Res. 2001;89(11):1073-1080.
doi - Slater SC, Ramnath RD, Uttridge K, Saleem MA, Cahill PA, Mathieson PW, Welsh GI, et al. Chronic exposure to laminar shear stress induces Kruppel-like factor 2 in glomerular endothelial cells and modulates interactions with co-cultured podocytes. Int J Biochem Cell Biol. 2012;44(9):1482-1490.
doi - Gracia-Sancho J, Russo L, Garcia-Caldero H, Garcia-Pagan JC, Garcia-Cardena G, Bosch J. Endothelial expression of transcription factor Kruppel-like factor 2 and its vasoprotective target genes in the normal and cirrhotic rat liver. Gut. 2011;60(4):517-524.
doi - Dekker RJ, Boon RA, Rondaij MG, Kragt A, Volger OL, Elderkamp YW, Meijers JC, et al. KLF2 provokes a gene expression pattern that establishes functional quiescent differentiation of the endothelium. Blood. 2006;107(11):4354-4363.
doi - Napoli C, de Nigris F, Williams-Ignarro S, Pignalosa O, Sica V, Ignarro LJ. Nitric oxide and atherosclerosis: an update. Nitric Oxide. 2006;15(4):265-279.
doi - Fledderus JO, van Thienen JV, Boon RA, Dekker RJ, Rohlena J, Volger OL, Bijnens AP, et al. Prolonged shear stress and KLF2 suppress constitutive proinflammatory transcription through inhibition of ATF2. Blood. 2007;109(10):4249-4257.
doi - Parmar KM, Larman HB, Dai G, Zhang Y, Wang ET, Moorthy SN, Kratz JR, et al. Integration of flow-dependent endothelial phenotypes by Kruppel-like factor 2. J Clin Invest. 2006;116(1):49-58.
doi pubmed pmc - SenBanerjee S, Lin Z, Atkins GB, Greif DM, Rao RM, Kumar A, Feinberg MW, et al. KLF2 Is a novel transcriptional regulator of endothelial proinflammatory activation. J Exp Med. 2004;199(10):1305-1315.
doi pubmed pmc - Eng E, Ballermann BJ. Diminished NF-kappaB activation and PDGF-B expression in glomerular endothelial cells subjected to chronic shear stress. Microvasc Res. 2003;65(3):137-144.
doi - Lin K, Hsu PP, Chen BP, Yuan S, Usami S, Shyy JY, Li YS, et al. Molecular mechanism of endothelial growth arrest by laminar shear stress. Proc Natl Acad Sci U S A. 2000;97(17):9385-9389.
doi pubmed pmc - Xu S, Xu Y, Yin M, Zhang S, Liu P, Koroleva M, Si S, et al. Flow-dependent epigenetic regulation of IGFBP5 expression by H3K27me3 contributes to endothelial anti-inflammatory effects. Theranostics. 2018;8(11):3007-3021.
doi pubmed pmc - Lin Z, Kumar A, SenBanerjee S, Staniszewski K, Parmar K, Vaughan DE, Gimbrone MA, Jr., et al. Kruppel-like factor 2 (KLF2) regulates endothelial thrombotic function. Circ Res. 2005;96(5):e48-57.
doi - Takada Y, Shinkai F, Kondo S, Yamamoto S, Tsuboi H, Korenaga R, Ando J. Fluid shear stress increases the expression of thrombomodulin by cultured human endothelial cells. Biochem Biophys Res Commun. 1994;205(2):1345-1352.
doi - Diamond SL, Eskin SG, McIntire LV. Fluid flow stimulates tissue plasminogen activator secretion by cultured human endothelial cells. Science. 1989;243(4897):1483-1485.
doi - Gaucher C, Devaux C, Boura C, Lacolley P, Stoltz JF, Menu P. In vitro impact of physiological shear stress on endothelial cells gene expression profile. Clin Hemorheol Microcirc. 2007;37(1-2):99-107
- Dewey CF, Jr., Bussolari SR, Gimbrone MA, Jr., Davies PF. The dynamic response of vascular endothelial cells to fluid shear stress. J Biomech Eng. 1981;103(3):177-185.
doi - Yoshizumi M, Abe J, Tsuchiya K, Berk BC, Tamaki T. Stress and vascular responses: atheroprotective effect of laminar fluid shear stress in endothelial cells: possible role of mitogen-activated protein kinases. J Pharmacol Sci. 2003;91(3):172-176.
doi - Yan C, Takahashi M, Okuda M, Lee JD, Berk BC. Fluid shear stress stimulates big mitogen-activated protein kinase 1 (BMK1) activity in endothelial cells. Dependence on tyrosine kinases and intracellular calcium. J Biol Chem. 1999;274(1):143-150.
doi - Tseng H, Peterson TE, Berk BC. Fluid shear stress stimulates mitogen-activated protein kinase in endothelial cells. Circ Res. 1995;77(5):869-878.
doi - Jo H, Sipos K, Go YM, Law R, Rong J, McDonald JM. Differential effect of shear stress on extracellular signal-regulated kinase and N-terminal Jun kinase in endothelial cells. Gi2- and Gbeta/gamma-dependent signaling pathways. J Biol Chem. 1997;272(2):1395-1401.
doi - Cunningham KS, Gotlieb AI. The role of shear stress in the pathogenesis of atherosclerosis. Lab Invest. 2005;85(1):9-23.
doi - Dardik A, Chen L, Frattini J, Asada H, Aziz F, Kudo FA, Sumpio BE. Differential effects of orbital and laminar shear stress on endothelial cells. J Vasc Surg. 2005;41(5):869-880.
doi - Akimoto S, Mitsumata M, Sasaguri T, Yoshida Y. Laminar shear stress inhibits vascular endothelial cell proliferation by inducing cyclin-dependent kinase inhibitor p21(Sdi1/Cip1/Waf1). Circ Res. 2000;86(2):185-190.
doi - Kayembe KN, Sasahara M, Hazama F. Cerebral aneurysms and variations in the circle of Willis. Stroke. 1984;15(5):846-850.
doi - Stehbens WE. Etiology of intracranial berry aneurysms. J Neurosurg. 1989;70(6):823-831.
doi - Gonzalez CF, Cho YI, Ortega HV, Moret J. Intracranial aneurysms: flow analysis of their origin and progression. AJNR Am J Neuroradiol. 1992;13(1):181-188.
pubmed pmc - Kondo S, Hashimoto N, Kikuchi H, Hazama F, Nagata I, Kataoka H. Cerebral aneurysms arising at nonbranching sites. An experimental Study. Stroke. 1997;28(2):398-403; discussion 403-394.
doi - Foutrakis GN, Yonas H, Sclabassi RJ. Saccular aneurysm formation in curved and bifurcating arteries. AJNR Am J Neuroradiol. 1999;20(7):1309-1317.
pubmed pmc - Matsuda M, Handa J, Saito A, Matsuda I, Kamijyo Y. Ruptured cerebral aneurysms associated with arterial occlusion. Surg Neurol. 1983;20(1):4-12.
doi - Salar G, Mingrino S. Ligature of the cervical carotid artery for the treatment of intracranial carotid aneurysms: complications and late results. Acta Neurochir (Wein). 1977;36:152.
- Yasargil MG. Microneurosurgery. Vol. 2. New York: Georg Thieme Verlag; 1984.
- Peerless SJ, Drake CG. Management of aneurysms of tahe posterior circulation. In: Youmans JR, editor. Neurological Surgery. Vol. 3. New York: Saunders; 1982. p. 1715-1763
- Steiger HJ. Pathophysiology of development and rupture of cerebral aneurysms. Acta Neurochir Suppl (Wien). 1990;48:1-57
- Gao B, Baharoglu MI, Cohen AD, Malek AM. Y-stent coiling of basilar bifurcation aneurysms induces a dynamic angular vascular remodeling with alteration of the apical wall shear stress pattern. Neurosurgery. 2013;72(4):617-629; discussion 628-619.
doi - Meng H, Wang Z, Hoi Y, Gao L, Metaxa E, Swartz DD, Kolega J. Complex hemodynamics at the apex of an arterial bifurcation induces vascular remodeling resembling cerebral aneurysm initiation. Stroke. 2007;38(6):1924-1931.
doi pubmed pmc - Jamous MA, Nagahiro S, Kitazato KT, Satoh K, Satomi J. Vascular corrosion casts mirroring early morphological changes that lead to the formation of saccular cerebral aneurysm: an experimental study in rats. J Neurosurg. 2005;102(3):532-535.
doi - Fukuda S, Hashimoto N, Naritomi H, Nagata I, Nozaki K, Kondo S, Kurino M, et al. Prevention of rat cerebral aneurysm formation by inhibition of nitric oxide synthase. Circulation. 2000;101(21):2532-2538.
doi - Hashimoto N, Handa H, Nagata I, Hazama F. Experimentally induced cerebral aneurysms in rats: Part V. Relation of hemodynamics in the circle of Willis to formation of aneurysms. Surg Neurol. 1980;13(1):41-45
- Kim C, Kikuchi H, Hashimoto N, Hazama F, Kataoka H. Establishment of the experimental conditions for inducing saccular cerebral aneurysms in primates with special reference to hypertension. Acta Neurochir (Wien). 1989;96(3-4):132-136.
doi - Nagata I, Handa H, Hashimoto N, Hazama F. Experimentally induced cerebral aneurysms in rats: Part VI. Hypertension. Surg Neurol. 1980;14(6):477-479
- Kolega J, Gao L, Mandelbaum M, Mocco J, Siddiqui AH, Natarajan SK, Meng H. Cellular and molecular responses of the basilar terminus to hemodynamics during intracranial aneurysm initiation in a rabbit model. J Vasc Res. 2011;48(5):429-442.
doi pubmed pmc - Jamous MA, Nagahiro S, Kitazato KT, Tamura T, Aziz HA, Shono M, Satoh K. Endothelial injury and inflammatory response induced by hemodynamic changes preceding intracranial aneurysm formation: experimental study in rats. J Neurosurg. 2007;107(2):405-411.
doi - Dolan JM, Sim FJ, Meng H, Kolega J. Endothelial cells express a unique transcriptional profile under very high wall shear stress known to induce expansive arterial remodeling. Am J Physiol Cell Physiol. 2012;302(8):C1109-1118.
doi pubmed pmc - Hoi Y, Gao L, Tremmel M, Paluch RA, Siddiqui AH, Meng H, Mocco J. In vivo assessment of rapid cerebrovascular morphological adaptation following acute blood flow increase. J Neurosurg. 2008;109(6):1141-1147.
doi pubmed pmc - Wang Z, Kolega J, Hoi Y, Gao L, Swartz DD, Levy EI, Mocco J, et al. Molecular alterations associated with aneurysmal remodeling are localized in the high hemodynamic stress region of a created carotid bifurcation. Neurosurgery. 2009;65(1):169-177; discussion 177-168.
doi pubmed pmc - Pawlowska E, Szczepanska J, Wisniewski K, Tokarz P, Jaskolski DJ, Blasiak J. NF-kappaB-Mediated Inflammation in the Pathogenesis of Intracranial Aneurysm and Subarachnoid Hemorrhage. Does Autophagy Play a Role? Int J Mol Sci. 2018;19(4):1245.
doi pubmed pmc - Taylor BES, Appelboom G, Zilinyi R, Goodman A, Chapel D, LoPresti M, et al. Role of the complement cascade in cerebral aneurysm formation, growth, and rupture. Neuroimmunol Neuroinflamm. 2015;2:93-101.
- Shi ZD, Tarbell JM. Fluid flow mechanotransduction in vascular smooth muscle cells and fibroblasts. Ann Biomed Eng. 2011;39(6):1608-1619.
doi pubmed pmc - Papadaki M, Ruef J, Nguyen KT, Li F, Patterson C, Eskin SG, McIntire LV, et al. Differential regulation of protease activated receptor-1 and tissue plasminogen activator expression by shear stress in vascular smooth muscle cells. Circ Res. 1998;83(10):1027-1034.
doi - Fukuda M, Aoki T. Molecular basis for intracranial aneurysm formation. Acta Neurochir Suppl. 2015;120:13-15.
doi - Kulcsar Z, Ugron A, Marosfoi M, Berentei Z, Paal G, Szikora I. Hemodynamics of cerebral aneurysm initiation: the role of wall shear stress and spatial wall shear stress gradient. AJNR Am J Neuroradiol. 2011;32(3):587-594.
doi pubmed pmc - Metaxa E, Tremmel M, Natarajan SK, Xiang J, Paluch RA, Mandelbaum M, Siddiqui AH, et al. Characterization of critical hemodynamics contributing to aneurysmal remodeling at the basilar terminus in a rabbit model. Stroke. 2010;41(8):1774-1782.
doi pubmed pmc - Aoki T, Nishimura M, Matsuoka T, Yamamoto K, Furuyashiki T, Kataoka H, Kitaoka S, et al. PGE(2) -EP(2) signalling in endothelium is activated by haemodynamic stress and induces cerebral aneurysm through an amplifying loop via NF-kappaB. Br J Pharmacol. 2011;163(6):1237-1249.
doi pubmed pmc - Orr AW, Sanders JM, Bevard M, Coleman E, Sarembock IJ, Schwartz MA. The subendothelial extracellular matrix modulates NF-kappaB activation by flow: a potential role in atherosclerosis. J Cell Biol. 2005;169(1):191-202.
doi pubmed pmc - Lan Q, Mercurius KO, Davies PF. Stimulation of transcription factors NF kappa B and AP1 in endothelial cells subjected to shear stress. Biochem Biophys Res Commun. 1994;201(2):950-956.
doi - Khachigian LM, Resnick N, Gimbrone MA, Jr., Collins T. Nuclear factor-kappa B interacts functionally with the platelet-derived growth factor B-chain shear-stress response element in vascular endothelial cells exposed to fluid shear stress. J Clin Invest. 1995;96(2):1169-1175.
doi pubmed pmc - Ballermann BJ, Dardik A, Eng E, Liu A. Shear stress and the endothelium. Kidney Int Suppl. 1998;67:S100-108.
doi - Davis ME, Grumbach IM, Fukai T, Cutchins A, Harrison DG. Shear stress regulates endothelial nitric-oxide synthase promoter activity through nuclear factor kappaB binding. J Biol Chem. 2004;279(1):163-168.
doi - Shiraya S, Miwa K, Aoki M, Miyake T, Oishi M, Kataoka K, Ohgi S, et al. Hypertension accelerated experimental abdominal aortic aneurysm through upregulation of nuclear factor kappaB and Ets. Hypertension. 2006;48(4):628-636.
doi - Bhullar IS, Li YS, Miao H, Zandi E, Kim M, Shyy JY, Chien S. Fluid shear stress activation of IkappaB kinase is integrin-dependent. J Biol Chem. 1998;273(46):30544-30549.
doi - Schneider A, Martin-Villalba A, Weih F, Vogel J, Wirth T, Schwaninger M. NF-kappaB is activated and promotes cell death in focal cerebral ischemia. Nat Med. 1999;5(5):554-559.
doi - Aoki T, Kataoka H, Ishibashi R, Nozaki K, Hashimoto N. Simvastatin suppresses the progression of experimentally induced cerebral aneurysms in rats. Stroke. 2008;39(4):1276-1285.
doi - Aoki T, Kataoka H, Ishibashi R, Nozaki K, Hashimoto N. Nifedipine inhibits the progression of an experimentally induced cerebral aneurysm in rats with associated down-regulation of NF-kappa B transcriptional activity. Curr Neurovasc Res. 2008;5(1):37-45.
doi - Aoki T, Kataoka H, Ishibashi R, Nakagami H, Nozaki K, Morishita R, Hashimoto N. Pitavastatin suppresses formation and progression of cerebral aneurysms through inhibition of the nuclear factor kappaB pathway. Neurosurgery. 2009;64(2):357-365; discussion 365-356.
doi - Lee JY, Kim JS, Kim JM, Kim N, Jung HC, Song IS. Simvastatin inhibits NF-kappaB signaling in intestinal epithelial cells and ameliorates acute murine colitis. Int Immunopharmacol. 2007;7(2):241-248.
doi - Cebral JR, Detmer F, Chung BJ, Choque-Velasquez J, Rezai B, Lehto H, Tulamo R, et al. Local hemodynamic conditions associated with focal changes in the intracranial aneurysm wall. AJNR Am J Neuroradiol. 2019;40(3):510-516.
doi pubmed pmc - Chung BJ, Mut F, Putman CM, Hamzei-Sichani F, Brinjikji W, Kallmes D, Jimenez CM, et al. Identification of hostile hemodynamics and geometries of cerebral aneurysms: a case-control study. AJNR Am J Neuroradiol. 2018;39(10):1860-1866.
doi pubmed pmc - Watton PN, Raberger NB, Holzapfel GA, Ventikos Y. Coupling the hemodynamic environment to the evolution of cerebral aneurysms: computational framework and numerical examples. J Biomech Eng. 2009;131(10):101003.
doi - Tateshima S, Murayama Y, Villablanca JP, Morino T, Nomura K, Tanishita K, Vinuela F. In vitro measurement of fluid-induced wall shear stress in unruptured cerebral aneurysms harboring blebs. Stroke. 2003;34(1):187-192.
doi - Kadasi LM, Dent WC, Malek AM. Colocalization of thin-walled dome regions with low hemodynamic wall shear stress in unruptured cerebral aneurysms. J Neurosurg. 2013;119(1):172-179.
doi - Boussel L, Rayz V, McCulloch C, Martin A, Acevedo-Bolton G, Lawton M, Higashida R, et al. Aneurysm growth occurs at region of low wall shear stress: patient-specific correlation of hemodynamics and growth in a longitudinal study. Stroke. 2008;39(11):2997-3002.
doi pubmed pmc - Skodvin TO, Johnsen LH, Gjertsen O, Isaksen JG, Sorteberg A. Cerebral aneurysm morphology before and after rupture: nationwide case series of 29 aneurysms. Stroke. 2017;48(4):880-886.
doi - Turjman AS, Turjman F, Edelman ER. Role of fluid dynamics and inflammation in intracranial aneurysm formation. Circulation. 2014;129(3):373-382.
doi pubmed pmc - Ross R, Glomset JA. The pathogenesis of atherosclerosis (first of two parts). N Engl J Med. 1976;295(7):369-377.
doi - Meng H, Tutino VM, Xiang J, Siddiqui A. High WSS or low WSS? Complex interactions of hemodynamics with intracranial aneurysm initiation, growth, and rupture: toward a unifying hypothesis. AJNR Am J Neuroradiol. 2014;35(7):1254-1262.
doi pubmed pmc - Sakamoto N, Saito N, Han X, Ohashi T, Sato M. Effect of spatial gradient in fluid shear stress on morphological changes in endothelial cells in response to flow. Biochem Biophys Res Commun. 2010;395(2):264-269.
doi - Penn DL, Komotar RJ, Sander Connolly E. Hemodynamic mechanisms underlying cerebral aneurysm pathogenesis. J Clin Neurosci. 2011;18(11):1435-1438.
doi - Griffith TM. Modulation of blood flow and tissue perfusion by endothelium-derived relaxing factor. Exp Physiol. 1994;79(6):873-913.
doi - Liepsch DW. Flow in tubes and arteries—a comparison. Biorheology. 1986;23(4):395-433.
doi - Moncada S, Plamer RMJ, Higgs EA. Nitric oxide: physiology, pathology and pharmacology. Pharmacol Rev. 1991;43:109-142.
- Moritake K, Handa H, Hayashi K, Sato M. Experimental studies on intracranial aneurysms (a preliminary report): some biomechanical considerations on the wall structures of intracranial aneurysms and experimentally produced aneurysms. No Shinkei Sheka. 1973;1:115-123.
- Walpola PL, Gotlieb AI, Cybulsky MI, Langille BL. Expression of ICAM-1 and VCAM-1 and monocyte adherence in arteries exposed to altered shear stress. Arterioscler Thromb Vasc Biol. 1995;15(1):2-10.
doi - Guzman RJ, Abe K, Zarins CK. Flow-induced arterial enlargement is inhibited by suppression of nitric oxide synthase activity in vivo. Surgery. 1997;122(2):273-279; discussion 279-280.
doi - Hara A, Yoshimi N, Mori H. Evidence for apoptosis in human intracranial aneurysms. Neurol Res. 1998;20(2):127-130.
doi - Sho E, Sho M, Singh TM, Xu C, Zarins CK, Masuda H. Blood flow decrease induces apoptosis of endothelial cells in previously dilated arteries resulting from chronic high blood flow. Arterioscler Thromb Vasc Biol. 2001;21(7):1139-1145.
doi - Caro CG, Fitz-Gerald JM, Schroter RC. Atheroma and arterial wall shear. Observation, correlation and proposal of a shear dependent mass transfer mechanism for atherogenesis. Proc R Soc Lond B Biol Sci. 1971;177(1046):109-159.
doi - Glagov S, Zarins C, Giddens DP, Ku DN. Hemodynamics and atherosclerosis. Insights and perspectives gained from studies of human arteries. Arch Pathol Lab Med. 1988;112(10):1018-1031
- Zarins CK, Giddens DP, Bharadvaj BK, Sottiurai VS, Mabon RF, Glagov S. Carotid bifurcation atherosclerosis. Quantitative correlation of plaque localization with flow velocity profiles and wall shear stress. Circ Res. 1983;53(4):502-514.
doi - Marchese E, Vignati A, Albanese A, Nucci CG, Sabatino G, Tirpakova B, Lofrese G, et al. Comparative evaluation of genome-wide gene expression profiles in ruptured and unruptured human intracranial aneurysms. J Biol Regul Homeost Agents. 2010;24(2):185-195
- Pentimalli L, Modesti A, Vignati A, Marchese E, Albanese A, Di Rocco F, Coletti A, et al. Role of apoptosis in intracranial aneurysm rupture. J Neurosurg. 2004;101(6):1018-1025.
doi - Malek AM, Alper SL, Izumo S. Hemodynamic shear stress and its role in atherosclerosis. JAMA. 1999;282(21):2035-2042.
doi - Qiu T, Jin G, Xing H, Lu H. Association between hemodynamics, morphology, and rupture risk of intracranial aneurysms: a computational fluid modeling study. Neurol Sci. 2017;38(6):1009-1018.
doi pubmed pmc - Galis ZS, Khatri JJ. Matrix metalloproteinases in vascular remodeling and atherogenesis: the good, the bad, and the ugly. Circ Res. 2002;90(3):251-262
- Lu D, Kassab GS. Role of shear stress and stretch in vascular mechanobiology. J R Soc Interface. 2011;8(63):1379-1385.
doi pubmed pmc - Zhou G, Zhu Y, Yin Y, Su M, Li M. Association of wall shear stress with intracranial aneurysm rupture: systematic review and meta-analysis. Sci Rep. 2017;7(1):5331.
doi pubmed pmc - Chiu JJ, Chien S. Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol Rev. 2011;91(1):327-387.
doi pubmed pmc - Sawyer DM, Pace LA, Pascale CL, Kutchin AC, O'Neill BE, Starke RM, Dumont AS. Lymphocytes influence intracranial aneurysm formation and rupture: role of extracellular matrix remodeling and phenotypic modulation of vascular smooth muscle cells. J Neuroinflammation. 2016;13(1):185.
doi pubmed pmc - Kataoka K, Taneda M, Asai T, Kinoshita A, Ito M, Kuroda R. Structural fragility and inflammatory response of ruptured cerebral aneurysms. A comparative study between ruptured and unruptured cerebral aneurysms. Stroke. 1999;30(7):1396-1401.
doi - Nuki Y, Matsumoto MM, Tsang E, Young WL, van Rooijen N, Kurihara C, Hashimoto T. Roles of macrophages in flow-induced outward vascular remodeling. J Cereb Blood Flow Metab. 2009;29(3):495-503.
doi pubmed pmc - Aoki T, Kataoka H, Morimoto M, Nozaki K, Hashimoto N. Macrophage-derived matrix metalloproteinase-2 and -9 promote the progression of cerebral aneurysms in rats. Stroke. 2007;38(1):162-169.
doi - Kanematsu Y, Kanematsu M, Kurihara C, Tada Y, Tsou TL, van Rooijen N, Lawton MT, et al. Critical roles of macrophages in the formation of intracranial aneurysm. Stroke. 2011;42(1):173-178.
doi pubmed pmc - Kurki MI, Hakkinen SK, Frosen J, Tulamo R, von und zu Fraunberg M, Wong G, Tromp G, et al. Upregulated signaling pathways in ruptured human saccular intracranial aneurysm wall: an emerging regulative role of Toll-like receptor signaling and nuclear factor-kappaB, hypoxia-inducible factor-1A, and ETS transcription factors. Neurosurgery. 2011;68(6):1667-1675; discussion 1675-1666.
doi - Hwang J, Ing MH, Salazar A, Lassegue B, Griendling K, Navab M, Sevanian A, et al. Pulsatile versus oscillatory shear stress regulates NADPH oxidase subunit expression: implication for native LDL oxidation. Circ Res. 2003;93(12):1225-1232.
doi pubmed pmc - Arnal JF, Dinh-Xuan AT, Pueyo M, Darblade B, Rami J. Endothelium-derived nitric oxide and vascular physiology and pathology. Cell Mol Life Sci. 1999;55(8-9):1078-1087.
doi - Tamura T, Jamous MA, Kitazato KT, Yagi K, Tada Y, Uno M, Nagahiro S. Endothelial damage due to impaired nitric oxide bioavailability triggers cerebral aneurysm formation in female rats. J Hypertens. 2009;27(6):1284-1292.
doi
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Neurology Research is published by Elmer Press Inc.