

| Journal of Neurology Research, ISSN 1923-2845 print, 1923-2853 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Neurol Res and Elmer Press Inc |
| Journal website http://www.neurores.org |
Review
Volume 3, Number 1, February 2013, pages 1-11
Patient Care and Treatment in Amyotrophic Lateral Sclerosis
Ken Ikedaa, b, Osamu Kanoa, Kiyokazu Kawabea, Yasuo Iwasakia
aDepartment of Neurology, Toho University Omori Medical Center, Tokyo, Japan
bCorresponding author: Ken Ikeda, Department of Neurology, Toho University Omori Medical Center, 6-11-1, Omorinishi, Otaku, Tokyo, 143-8541, Japan
Manuscript accepted for publication January 30, 2013
Short title: Patient Care and Treatment
doi: https://doi.org/10.4021/jnr176w
| Abstract | ▴Top |
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by the loss of upper and lower motor neurons, leading to limb paralysis and respiratory failure. ALS is considered as an incurable disease. However, the management has considerably changed for recent years. Currently, four crucial sections of practice and treatment have been recommended for prolonging the survival of ALS patients: 1) symptomatic treatment; 2) respiratory management; 3) nutritional management; and 4) disease modifying medication. Although many clinical symptoms occur during the disease course, those symptoms are treatable in ALS patients. All efforts should be made to improve quality of life and to assist in keeping the patient’s autonomy. Advanced directives on end of life care, respiratory and nutritional management during late stages of life are important issues. This review article introduces current treatment, including symptomatic treatments, ventilation, nutrition, disease modifying medication or condition in ALS patients, including riluzole, serum levels of cholesterol, urate and ferritin, and clinical therapeutic trials.
Keywords: Amyotrophic lateral sclerosis; Multidisciplinary care; Symptomatic treatment; Ventilation; Nutrition; Disease modifying medication
| Introduction | ▴Top |
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by the loss of motor neurons in the cerebral cortex, brainstem and spinal cord, leading to limb paralysis and respiratory failure. Patient care and therapy of ALS has considerably amended over the past two decades. Recently, three effective treatments have been recommended for prolonging survival in ALS patients: multidisciplinary team care, mechanical ventilation and gastrostomy tubes [1, 2]. Although ALS is considered as an incurable disease, many symptoms occurring during the disease course are treatable. All efforts should be made to improve quality of life and to assist in keeping the patient’s autonomy. Advanced directives on end of life care, respiratory and nutritional management during late stages of life are important issues. ALS patients and their relatives need to discuss at the earliest opportunity that they are willing to do those management. They often suffer from depression, feelings of hopelessness and anxiety regarding end-of-life problems following the diagnosis of ALS or with the disease progresses [3, 4]. Therefore, psychological counsel and palliative care should be offered to the patients and relatives early [1, 2, 5, 6]. This review introduces current treatment in ALS patients at the following four divisions: symptomatic treatment; ventilation; nutrition; and disease modifying medication.
Symptomatic treatment
ALS patients develop numerous manifestations and problems during the disease course. Symptomatic treatment aims to improve quality of life in ALS patients and their caregivers. Main symptoms of ALS patients and those medications are summarized in Table 1. Physicians need to monitor and care for these symptoms from the early stage of ALS.
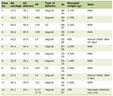 Click to view | Table 1. Symptomatic Treatments in ALS Patients |
Respiratory management
Respiratory insufficiency occurs commonly in ALS patients and it is a major cause of mortality. Clinical symptoms due to respiratory muscle weakness include dyspnoea on exertion or talking, orthopnea, disturbed sleep, excessive daytime somnolence, morning headaches, fatigue, anorexia, depression, poor concentration, vivid nightmares and nocturia. Clinical examination reveals tachypnea, use of accessory breathing muscles, paradoxical movement of the abdomen, weak cough and rarely papilledema [5, 7]. The forced vital capacity (FVC) is the most widely used for measurement of respiratory function in ALS [8], and it was a significant predictor of survival [9]. Measurement of the Sniff nasal inspiratory pressure (SNIP) is a useful parameter for the diaphragmatic strength. SNIP is probably more accurate than vital capacity, although both measurements are unable to estimate respiratory function accurately in patients with bulbar palsy. Published guidelines for respiratory care were based on clinical experience, expert opinion, and observational research [7, 10, 11]. Recently, the American Academy of Neurology (AAN) ALS Practice Parameter recommends consideration of noninvasive ventilation (NIV) when patients have orthopnea, SNIP < 40 cm or maximal inspiratory pressure < -60 cm, abnormal nocturnal oximetry or FVC < 50% (Fig. 1) [1]. Otherwise, patients with FVC ≥ 70% occasionally develop respiratory failure. Therefore, FVC ≤ 75% is probably more appropriate for earlier monitoring of respiratory symptoms [2-14]. SNIP < 25 cm H2O is highly predictive of respiratory failure [15]. Overnight oximetry can detect episodes of nocturnal hypoventilation [16]. Ventilator support is usually provided by NIV without tracheostomy. Bi-level positive pressure devices are commonly used in NIV, whereas continuous positive pressure ventilation is not usually helpful [17]. NIV initially applied for intermittent nocturnal support to alleviate symptoms due to nocturnal hypoventilation. When respiratory function worsens, patients tend to require daytime support and eventually continuous support. Observational studies and a recent randomised controlled trial involving 92 ALS patients show that NIV improves survival and quality of life [18, 19]. In patients with severe bulbar impairment, NIV improves sleep-related symptoms, but is unlikely to confer a large survival advantage [19]. Finally, AAN Practice Parameter of ALS care recommends that NIV should be considered to treat respiratory insufficiency in ALS patients for lengthening survival and slowing FVC decline rate [1].
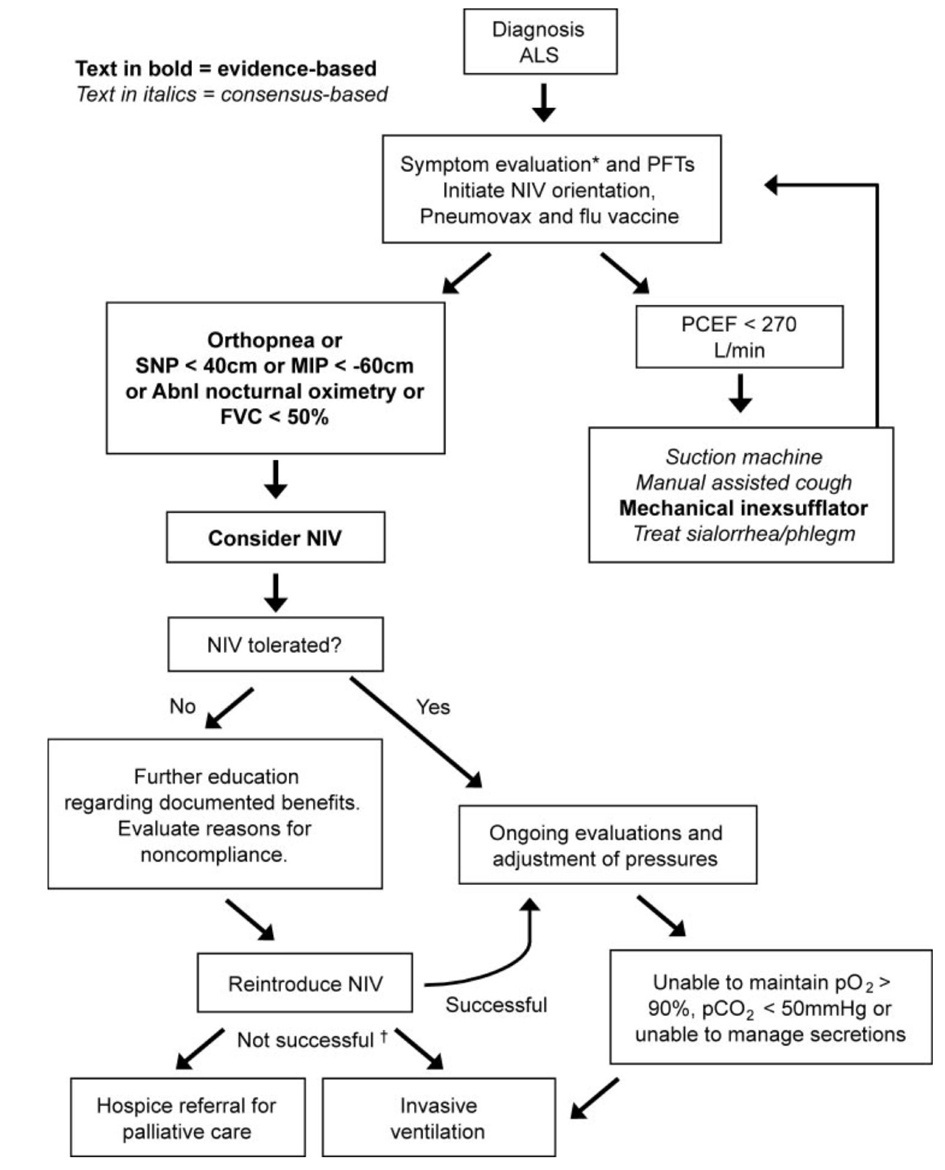 Click for large image | Figure 1. Respiratory management algorithm by AAN ALS Practice Parameter [1]. PFT: pulmonary function tests. PCEF: peak cough expiratory flow. NIV: noninvasive ventilation. SNP: sniff nasal pressure. MIP: maximal inspiratory pressure. FVC: forced vital capacity (supine or erect). Abnl. nocturnal oximetry =pO2 < 4% from baseline. *Symptoms suggestive of nocturnal hypoventilation: frequent arousals, morning headaches, excessive daytime sleepiness, vivid dreams. †If NIV is not tolerated or accepted in the setting of advancing respiratory compromise, consider invasive ventilation or referral to hospice. |
Nutritional management
In ALS patients, adequate nutrition is restricted insidiously and nutritional conditions worsen progressively. Dysphagia is a usual symptom of ALS and leads to increased risk of aspiration, malnutrition, weight loss and dehydration. Malnutrition and dehydration can also occur in patients with severe upper limb weakness because they have difficulties in meal preparation or prolonged meal times. ALS patients are reported to have a hypermetabolic state. They require increased calorie intake [20, 21]. Early management of dysphagia contains dietary advice, modification of food consistency (blending solid, adding thickening agents to liquids) and patient education on special swallowing techniques, such as supraglottic swallowing and postural changes (‘Chin tuck maneuver’) [5, 22]. Most guidelines address that supplementary enteral feeding should be considered when body weight loss (BMI) > 10% of the pre-diagnostic or baseline weight [5, 22]. Available enteric feedings include percutaneous endoscopic gastrostomy (PEG), percutaneous radiologic gastrostomy (PRG), radiologically inserted gastrostomy (RIG), and nasogastric tube (NGT). For therapeutic strategies to keep oral nutritional intake, PEG or RIG may be needed as an alternative route for delivering nutrition (Fig. 2). Physicians should emphasize to patients that PEG does not eliminate oral feeding. PEG is a convenient method for administering medication and fluid, and stabilizing body weight [23]. PEG is the standard procedure for enteral feeding, although the procedure requires mild sedation and has inference in patients with respiratory disturbance. To lower the risk, several studies recommend that PEG should be performed at VC > 50% [1, 5, 24]. Although it may be possible to insert PEG with NIV assistance, PRG/RIG insertion is a better alternative in these patients [25-27]. NGT is a relatively non-invasive option, but its long-term use is limited by discomfort and frequent replacement. Therefore, NGT should only be considered in patients who cannot undergo PEG or RIG insertion.
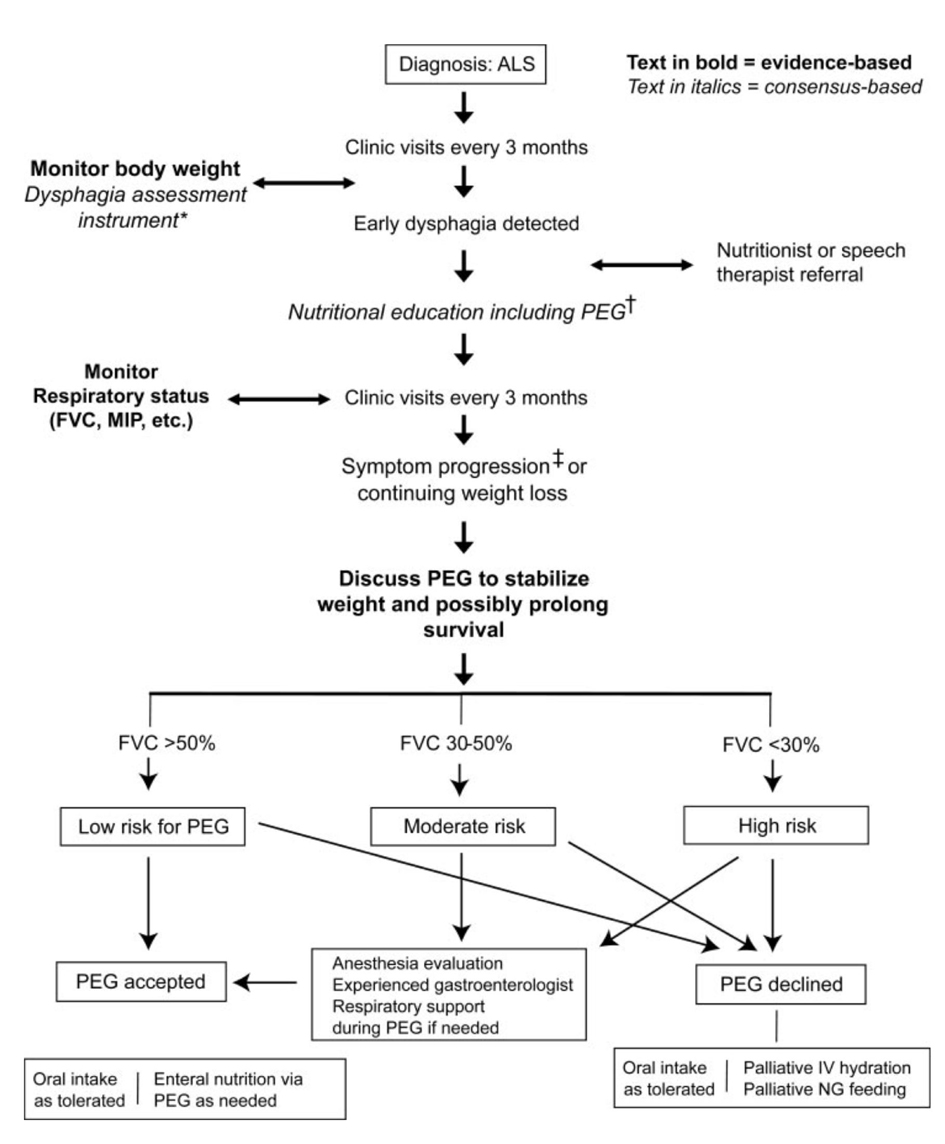 Click for large image | Figure 2. Nutrition management algorithm by AAN ALS Practice Parameter [1]. *e.g., Bulbar questions in the Amyotrophic Lateral Sclerosis Functional Rating Scale, or other instrument.†Prolonged meal time; ending meal prematurely because of fatigue; accelerated weight loss due to poor caloric intake; family concern about feeding difficulties. ‡Percutaneous endoscopic gastrostomy: rule out contraindication. |
Disease modifying treatment or condition
Riluzole
Despite many clinical trials and various advances in the understanding of ALS, there has been little successful treatment for disease modifying or neuroprotective agents. Riluzole is the only approved drug that has been shown to have a modest effect on prolonging survival in ALS patients [28-33]. The mechanism of riluzole is though to include interference with N-methyl-D-aspartate receptors, stabilization of the inactivated state of voltage-dependent sodium channels, inhibition of glutamate release from presynaptic terminals and increasing of extracellular glutamate uptake [34]. The AAN practice advisory recommended riluzole at 100 mg/day to extend survival in ALS patients clinically probable or definite El Escorial ALS who had symptoms less than 5 years, FVC > 60% and non-tracheostomy [35]. Cochrane Collaboration meta-analysis showed that the absolute risk reduction with the 100 mg dose at 12 months was 9%, with the numbers needed-to treat to delay one death after 12 months is 11 [36]. The drug is generally well tolerated and the most common side effects contain asthenia, nausea, gastrointestinal upset and liver dysfunction. Therefore liver function should be regularly monitored during therapy [37].
Serum levels of lipid, urate and ferritin
Previous studies have reported distinct serological profiles of lipid, urate and ferritin in Western and Japanese patients with ALS. The main previous studies of lipid, urate and ferritin levels in blood samples of ALS patients are summarized in Table 2.
Click to view | Table 2. Main Previous Linked Studies Between Disease Progression and Lipid, Urate and Ferritin in Sera or Plasma of ALS Patients |
1) Serum lipid levels
Four large European studies showed that serum lipid levels were related to better respiratory function or longer survival in ALS patients [38-41]. In a recent cross-sectional study, baseline serum levels of total cholesterol and low-density lipoprotein-cholesterol were independent predictors for rapid worsening of ALS-Functional Rating Scale-Revised (ALS-FRS) and FVC in Japanese patients [42]. In contrast, two studies showed no significant longer survival in ALS patients with hyperlipidemia [43, 44].
Otherwise, there is no consensus of statin use. Lipid-lowering treatment has been debated in occurrence and survival of ALS patients. Some database studies showed an apparent increased developmental risk of ALS for patients taking statins, whereas others suggested no association between ALS onset and statin use [45]. Some cohort studies exhibited an increased progression rate in ALS patients taking statins [46]. In contrast, others described that statins had no significant effects on survival [47]. At least, statins may be used in ALS patients with slowly progression and multiple cardiovascular disease risk factors.
2) Serum urate levels
A previous Israeli study described lower urate levels in sera of ALS patients compared to controls matched for age, sex and body mass index. The univariate analysis showed that relative reduction of serum urate levels was inversely correlated with ALS-FRS decline rate [48]. A recent Italian study described decreased levels of urate in sera of ALS patients [49]. ALS patients with long disease duration had significantly lower serum urate levels compared to controls. Greater reductions of serum urate levels were observed in patients with bulbar onset compared to patients with limb onset [49]. Our previous study indicated that baseline serum urate levels were an independent factor for rapid worsening of ALS-FRS or FVC [42]. Thus, higher urate levels could have favorable effects on both nutrition and disease progression in ALS patients.
3) Serum ferritin levels
Serum ferritin levels are well known as a marker for iron storage in humans. In a previous study, increased serum ferritin levels over one year were correlated with rapid progression of muscle weakness and shorter survival [50]. Another study also suggested higher serum ferritin levels in ALS patients, and a significant association between serum ferritin levels and rapid worsening of ALS-FRS and FVC [42].
Metabolic and nutritional conditions of lipid, urate, iron and BMI could contribute to disease progression in ALS patients [38-43, 48-50]. Finally, further studies investigating high nutrition diets and iron chelating are warranted for the treatment of ALS.
Phase II or III clinical trials
More than 100 neuroprotective or neurotrophic agents have been treated in ALS patients [51-75]. Main phase II or III human clinical trials are listed in Table 3. Most of those clinical trials have shown inconclusive evidence or demonstrate no beneficial effects on routine clinical practice.
Click to view | Table 3. Main Previous Clinical Trials of Phase II or III in ALS Patients |
Future hopeful treatment
As gene therapy approach to deliver neurotrophic factors directly to motor neurons, genetically engineered adeno-associated viruses (AAV) expressing neurotrophic factor genes was applied. Those AAV evaluated in mutant superoxide dismutase 1-transgenic mice and some promising results were reported [76]. However, human clinical studies are not yet underway. Another novel approach is the use of autologous stem cell transplantation. So far there have been no convincing results in human studies [77, 78]. Recent discovery show that induced pluripotent stem (iPS) cells generated from ALS patients are differentiated into motor neurons [79]. Production and therapeutic application of ALS-targeted iPS cells are strongly expected in the near future.
| Conclusion | ▴Top |
As multidisciplinary care in the United States of America, ALS patients are cured by their neurologist together with a team of therapists, usually including speech therapists, physical and occupational therapists, respiratory therapists, a nutritionist and a social worker [1, 2]. Cooperation between physicians and medical experts is required for better management and medication in ALS patients. According to the medical insurance system and national circumstances in each country, medical and social supports are needed for care of ALS patients.
Conflict of Interest
The authors state that they have no conflict of interest (COI).
| References | ▴Top |
- Miller RG, Jackson CE, Kasarskis EJ, England JD, Forshew D, Johnston W, Kalra S, et al. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: drug, nutritional, and respiratory therapies (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2009;73(15):1218-1226.
doi pubmed - Miller RG, Jackson CE, Kasarskis EJ, England JD, Forshew D, Johnston W, Kalra S, et al. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: multidisciplinary care, symptom management, and cognitive/behavioral impairment (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2009;73(15):1227-1233.
doi pubmed - Averill AJ, Kasarskis EJ, Segerstrom SC. Psychological health in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2007;8(4):243-254.
doi pubmed - Wicks P, Abrahams S, Masi D, Hejda-Forde S, Leigh PN, Goldstein LH. Prevalence of depression in a 12-month consecutive sample of patients with ALS. Eur J Neurol. 2007;14(9):993-1001.
doi pubmed - Leigh PN, Abrahams S, Al-Chalabi A, Ampong MA, Goldstein LH, Johnson J, Lyall R, et al. The management of motor neurone disease. J Neurol Neurosurg Psychiatry. 2003;74(Suppl 4):iv32-iv47.
doi pubmed - Mitsumoto H, Rabkin JG. Palliative care for patients with amyotrophic lateral sclerosis: "prepare for the worst and hope for the best". JAMA. 2007;298(2):207-216.
doi pubmed - Heffernan C, Jenkinson C, Holmes T, Macleod H, Kinnear W, Oliver D, Leigh N, et al. Management of respiration in MND/ALS patients: an evidence based review. Amyotroph Lateral Scler. 2006;7(1):5-15.
doi pubmed - Melo J, Homma A, Iturriaga E, Frierson L, Amato A, Anzueto A, Jackson C. Pulmonary evaluation and prevalence of non-invasive ventilation in patients with amyotrophic lateral sclerosis: a multicenter survey and proposal of a pulmonary protocol. J Neurol Sci. 1999;169(1-2):114-117.
doi - Czaplinski A, Yen AA, Appel SH. Forced vital capacity (FVC) as an indicator of survival and disease progression in an ALS clinic population. J Neurol Neurosurg Psychiatry. 2006;77(3):390-392.
doi pubmed - Miller RG, Rosenberg JA, Gelinas DF, Mitsumoto H, Newman D, Sufit R, Borasio GD, et al. Practice parameter: the care of the patient with amyotrophic lateral sclerosis (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology: ALS Practice Parameters Task Force. Neurology. 1999;52(7):1311-1323.
doi pubmed - Lechtzin N, Rothstein J, Clawson L, Diette GB, Wiener CM. Amyotrophic lateral sclerosis: evaluation and treatment of respiratory impairment. Amyotroph Lateral Scler Other Motor Neuron Disord. 2002;3(1):5-13.
doi pubmed - Lechtzin N, Wiener CM, Shade DM, Clawson L, Diette GB. Spirometry in the supine position improves the detection of diaphragmatic weakness in patients with amyotrophic lateral sclerosis. Chest. 2002;121(2):436-442.
doi pubmed - Gruis KL, Brown DL, Schoennemann A, Zebarah VA, Feldman EL. Predictors of noninvasive ventilation tolerance in patients with amyotrophic lateral sclerosis. Muscle Nerve. 2005;32(6):808-811.
doi pubmed - Schmidt EP, Drachman DB, Wiener CM, Clawson L, Kimball R, Lechtzin N. Pulmonary predictors of survival in amyotrophic lateral sclerosis: use in clinical trial design. Muscle Nerve. 2006;33(1):127-132.
doi pubmed - Lyall RA, Donaldson N, Polkey MI, Leigh PN, Moxham J. Respiratory muscle strength and ventilatory failure in amyotrophic lateral sclerosis. Brain. 2001;124(Pt 10):2000-2013.
doi pubmed - Elman LB, Siderowf AD, McCluskey LF. Nocturnal oximetry: utility in the respiratory management of amyotrophic lateral sclerosis. Am J Phys Med Rehabil. 2003;82(11):866-870.
doi pubmed - Radunovic A, Mitsumoto H, Leigh PN. Clinical care of patients with amyotrophic lateral sclerosis. Lancet Neurol. 2007;6(10):913-925.
doi - Bourke SC, Bullock RE, Williams TL, Shaw PJ, Gibson GJ. Noninvasive ventilation in ALS: indications and effect on quality of life. Neurology. 2003;61(2):171-177.
doi pubmed - Bourke SC, Tomlinson M, Williams TL, Bullock RE, Shaw PJ, Gibson GJ. Effects of non-invasive ventilation on survival and quality of life in patients with amyotrophic lateral sclerosis: a randomised controlled trial. Lancet Neurol. 2006;5(2):140-147.
doi - Desport JC, Preux PM, Magy L, Boirie Y, Vallat JM, Beaufrere B, Couratier P. Factors correlated with hypermetabolism in patients with amyotrophic lateral sclerosis. Am J Clin Nutr. 2001;74(3):328-334.
pubmed - Kasarskis EJ, Berryman S, Vanderleest JG, Schneider AR, McClain CJ. Nutritional status of patients with amyotrophic lateral sclerosis: relation to the proximity of death. Am J Clin Nutr. 1996;63(1):130-137.
pubmed - Andersen PM, Borasio GD, Dengler R, Hardiman O, Kollewe K, Leigh PN, Pradat PF, et al. EFNS task force on management of amyotrophic lateral sclerosis: guidelines for diagnosing and clinical care of patients and relatives. Eur J Neurol. 2005;12(12):921-938.
doi pubmed - Loser C, Aschl G, Hebuterne X, Mathus-Vliegen EM, Muscaritoli M, Niv Y, Rollins H, et al. ESPEN guidelines on artificial enteral nutrition—percutaneous endoscopic gastrostomy (PEG). Clin Nutr. 2005;24(5):848-861.
doi pubmed - Mathus-Vliegen LM, Louwerse LS, Merkus MP, Tytgat GN, Vianney de Jong JM. Percutaneous endoscopic gastrostomy in patients with amyotrophic lateral sclerosis and impaired pulmonary function. Gastrointest Endosc. 1994;40(4):463-469.
doi - Chio A, Galletti R, Finocchiaro C, Righi D, Ruffino MA, Calvo A, Di Vito N, et al. Percutaneous radiological gastrostomy: a safe and effective method of nutritional tube placement in advanced ALS. J Neurol Neurosurg Psychiatry. 2004;75(4):645-647.
doi pubmed - Heffernan C, Jenkinson C, Holmes T, Feder G, Kupfer R, Leigh PN, McGowan S, et al. Nutritional management in MND/ALS patients: an evidence based review. Amyotroph Lateral Scler Other Motor Neuron Disord. 2004;5(2):72-83.
doi pubmed - Shaw AS, Ampong MA, Rio A, McClure J, Leigh PN, Sidhu PS. Entristar skin-level gastrostomy tube: primary placement with radiologic guidance in patients with amyotrophic lateral sclerosis. Radiology. 2004;233(2):392-399.
doi pubmed - Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med. 1994;330(9):585-591.
doi pubmed - Lacomblez L, Bensimon G, Leigh PN, Guillet P, Powe L, Durrleman S, Delumeau JC, et al. A confirmatory dose-ranging study of riluzole in ALS. ALS/Riluzole Study Group-II. Neurology. 1996;47(6 Suppl 4):S242-250.
doi pubmed - Bensimon G, Lacomblez L, Delumeau JC, Bejuit R, Truffinet P, Meininger V. A study of riluzole in the treatment of advanced stage or elderly patients with amyotrophic lateral sclerosis. J Neurol. 2002;249(5):609-615.
doi pubmed - Meininger V, Lacomblez L, Salachas F. What has changed with riluzole? J Neurol. 2000;247(Suppl 6):VI/19-22.
- Mitchell JD, O'Brien M R, Joshi M. Audit of outcomes in motor neuron disease (MND) patients treated with riluzole. Amyotroph Lateral Scler. 2006;7(2):67-71.
doi pubmed - Turner MR, Parton MJ, Leigh PN. Clinical trials in ALS: an overview. Semin Neurol. 2001;21(2):167-175.
doi pubmed - Distad BJ, Meekins GD, Liou LL, Weiss MD, Carter GT, Miller RG. Drug therapy in amyotrophic lateral sclerosis. Phys Med Rehabil Clin N Am. 2008;19(3):633-651, xi-xii.
doi pubmed - Practice advisory on the treatment of amyotrophic lateral sclerosis with riluzole: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 1997;49(3):657-659.
doi - Miller RG, Mitchell JD, Lyon M, Moore DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev. 2007;(1):CD001447.
- Bensimon G, Doble A. The tolerability of riluzole in the treatment of patients with amyotrophic lateral sclerosis. Expert Opin Drug Saf. 2004;3(6):525-534.
doi - Dupuis L, Corcia P, Fergani A, Gonzalez De Aguilar JL, Bonnefont-Rousselot D, Bittar R, Seilhean D, et al. Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology. 2008;70(13):1004-1009.
doi pubmed - Chio A, Calvo A, Ilardi A, Cavallo E, Moglia C, Mutani R, Palmo A, et al. Lower serum lipid levels are related to respiratory impairment in patients with ALS. Neurology. 2009;73(20):1681-1685.
doi pubmed - Dorst J, Kuhnlein P, Hendrich C, Kassubek J, Sperfeld AD, Ludolph AC. Patients with elevated triglyceride and cholesterol serum levels have a prolonged survival in amyotrophic lateral sclerosis. J Neurol. 2011;258(4):613-617.
doi pubmed - Sutedja NA, van der Schouw YT, Fischer K, Sizoo EM, Huisman MH, Veldink JH, Van den Berg LH. Beneficial vascular risk profile is associated with amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2011;82(6):638-642.
doi pubmed - Ikeda K, Hirayama T, Takazawa T, Kawabe K, Iwasaki Y. Relationships between disease progression and serum levels of lipid, urate, creatinine and ferritin in Japanese patients with amyotrophic lateral sclerosis: a cross-sectional study. Intern Med. 2012;51(12):1501-1508.
doi pubmed - Paganoni S, Deng J, Jaffa M, Cudkowicz ME, Wills AM. Body mass index, not dyslipidemia, is an independent predictor of survival in amyotrophic lateral sclerosis. Muscle Nerve. 2011;44(1):20-24.
doi pubmed - Dedic SI, Stevic Z, Dedic V, Stojanovic VR, Milicev M, Lavrnic D. Is hyperlipidemia correlated with longer survival in patients with amyotrophic lateral sclerosis? Neurol Res. 2012;34(6):576-580.
doi pubmed - Sorensen HT, Lash TL. Statins and amyotrophic lateral sclerosis—the level of evidence for an association. J Intern Med. 2009;266(6):520-526.
doi pubmed - Zinman L, Sadeghi R, Gawel M, Patton D, Kiss A. Are statin medications safe in patients with ALS? Amyotroph Lateral Scler. 2008;9(4):223-228.
doi pubmed - Drory VE, Bronipolsky T, Artamonov I, Nefussy B. Influence of statins treatment on survival in patients with amyotrophic lateral sclerosis. J Neurol Sci. 2008;273(1-2):81-83.
doi pubmed - Keizman D, Ish-Shalom M, Berliner S, Maimon N, Vered Y, Artamonov I, Tsehori J, et al. Low uric acid levels in serum of patients with ALS: further evidence for oxidative stress? J Neurol Sci. 2009;285(1-2):95-99.
doi pubmed - Zoccolella S, Simone IL, Capozzo R, Tortelli R, Leo A, D'Errico E, Logroscino G. An exploratory study of serum urate levels in patients with amyotrophic lateral sclerosis. J Neurol. 2011;258(2):238-243.
doi pubmed - Qureshi M, Brown RH, Jr., Rogers JT, Cudkowicz ME. Serum ferritin and metal levels as risk factors for amyotrophic lateral sclerosis. Open Neurol J. 2008;2:51-54.
doi pubmed - Miller RG, Petajan JH, Bryan WW, Armon C, Barohn RJ, Goodpasture JC, Hoagland RJ, et al. A placebo-controlled trial of recombinant human ciliary neurotrophic (rhCNTF) factor in amyotrophic lateral sclerosis. rhCNTF ALS Study Group. Ann Neurol. 1996;39(2):256-260.
doi pubmed - A double-blind placebo-controlled clinical trial of subcutaneous recombinant human ciliary neurotrophic factor (rHCNTF) in amyotrophic lateral sclerosis. ALS CNTF Treatment Study Group. Neurology. 1996;46(5):1244-1249.
doi pubmed - Ochs G, Penn RD, York M, Giess R, Beck M, Tonn J, Haigh J, et al. A phase I/II trial of recombinant methionyl human brain derived neurotrophic factor administered by intrathecal infusion to patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1(3):201-206.
doi pubmed - Meininger V, Bensimon G, Bradley WR, Brooks B, Douillet P, Eisen AA, Lacomblez L, et al. Efficacy and safety of xaliproden in amyotrophic lateral sclerosis: results of two phase III trials. Amyotroph Lateral Scler Other Motor Neuron Disord. 2004;5(2):107-117.
doi pubmed - Mitchell JD, Wokke JH, Borasio GD. Recombinant human insulin-like growth factor I (rhIGF-I) for amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database Syst Rev. 20074):CD002064.
- Kaji R, Kodama M, Imamura A, Hashida T, Kohara N, Ishizu M, Inui K, et al. Effect of ultrahigh-dose methylcobalamin on compound muscle action potentials in amyotrophic lateral sclerosis: a double-blind controlled study. Muscle Nerve. 1998;21(12):1775-1778.
doi - Desnuelle C, Dib M, Garrel C, Favier A. A double-blind, placebo-controlled randomized clinical trial of alpha-tocopherol (vitamin E) in the treatment of amyotrophic lateral sclerosis. ALS riluzole-tocopherol Study Group. Amyotroph Lateral Scler Other Motor Neuron Disord. 2001;2(1):9-18.
doi pubmed - Graf M, Ecker D, Horowski R, Kramer B, Riederer P, Gerlach M, Hager C, et al. High dose vitamin E therapy in amyotrophic lateral sclerosis as add-on therapy to riluzole: results of a placebo-controlled double-blind study. J Neural Transm. 2005;112(5):649-660.
doi pubmed - Groeneveld GJ, Veldink JH, van der Tweel I, Kalmijn S, Beijer C, de Visser M, Wokke JH, et al. A randomized sequential trial of creatine in amyotrophic lateral sclerosis. Ann Neurol. 2003;53(4):437-445.
doi pubmed - Shefner JM, Cudkowicz ME, Schoenfeld D, Conrad T, Taft J, Chilton M, Urbinelli L, et al. A clinical trial of creatine in ALS. Neurology. 2004;63(9):1656-1661.
doi pubmed - Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, Jin L, et al. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433(7021):73-77.
doi pubmed - Traynor BJ, Bruijn L, Conwit R, Beal F, O'Neill G, Fagan SC, Cudkowicz ME. Neuroprotective agents for clinical trials in ALS: a systematic assessment. Neurology. 2006;67(1):20-27.
doi pubmed - Cudkowicz ME, Shefner JM, Schoenfeld DA, Zhang H, Andreasson KI, Rothstein JD, Drachman DB. Trial of celecoxib in amyotrophic lateral sclerosis. Ann Neurol. 2006;60(1):22-31.
doi pubmed - Yoshino H, Kimura A. Investigation of the therapeutic effects of edaravone, a free radical scavenger, on amyotrophic lateral sclerosis (Phase II study). Amyotroph Lateral Scler. 2006;7(4):241-245.
doi pubmed - Gordon PH, Moore DH, Miller RG, Florence JM, Verheijde JL, Doorish C, Hilton JF, et al. Efficacy of minocycline in patients with amyotrophic lateral sclerosis: a phase III randomised trial. Lancet Neurol. 2007;6(12):1045-1053.
doi - Miller R, Bradley W, Cudkowicz M, Hubble J, Meininger V, Mitsumoto H, Moore D, et al. Phase II/III randomized trial of TCH346 in patients with ALS. Neurology. 2007;69(8):776-784.
doi pubmed - Gribkoff VK, Bozik ME. KNS-760704 [(6R)-4,5,6,7-tetrahydro-N6-propyl-2, 6-benzothiazole-diamine dihydrochloride monohydrate] for the treatment of amyotrophic lateral sclerosis. CNS Neurosci Ther. 2008;14(3):215-226.
doi pubmed - Cudkowicz M, Bozik ME, Ingersoll EW, Miller R, Mitsumoto H, Shefner J, Moore DH, et al. The effects of dexpramipexole (KNS-760704) in individuals with amyotrophic lateral sclerosis. Nat Med. 2011;17(12):1652-1656.
doi pubmed - Lanka V, Wieland S, Barber J, Cudkowicz M. Arimoclomol: a potential therapy under development for ALS. Expert Opin Investig Drugs. 2009;18(12):1907-1918.
doi pubmed - Pascuzzi RM, Shefner J, Chappell AS, Bjerke JS, Tamura R, Chaudhry V, Clawson L, et al. A phase II trial of talampanel in subjects with amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2010;11(3):266-271.
doi pubmed - Chio A, Borghero G, Calvo A, Capasso M, Caponnetto C, Corbo M, Giannini F, et al. Lithium carbonate in amyotrophic lateral sclerosis: lack of efficacy in a dose-finding trial. Neurology. 2010;75(7):619-625.
doi pubmed - Clinical trial of tamoxifen [http://www.alsa.org/patient/drug.cfm?id=671].
- High-dose coenzyme Q10 shows no benefit in ALS. [http://www.als-mda.org/research/news/080423coenzyme.html].
- Teva provides update on glatiramer acetate 40 mg for amyotrophic lateral sclerosis (ALS) [http://www.medicalnewstoday.com/articles/100725.php].
- Shefner J, Cedarbaum JM, Cudkowicz ME, Maragakis N, Lee J, Jones D, Watson ML, et al. Safety, tolerability and pharmacodynamics of a skeletal muscle activator in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2012;13(5):430-438.
doi pubmed - Kaspar BK, Llado J, Sherkat N, Rothstein JD, Gage FH. Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science. 2003;301(5634):839-842.
doi pubmed - Mazzini L, Mareschi K, Ferrero I, Vassallo E, Oliveri G, Boccaletti R, Testa L, et al. Autologous mesenchymal stem cells: clinical applications in amyotrophic lateral sclerosis. Neurol Res. 2006;28(5):523-526.
doi pubmed - Mazzini L, Mareschi K, Ferrero I, Vassallo E, Oliveri G, Nasuelli N, Oggioni GD, et al. Stem cell treatment in Amyotrophic Lateral Sclerosis. J Neurol Sci. 2008;265(1-2):78-83.
doi pubmed - Dimos JT, Rodolfa KT, Niakan KK, Weisenthal LM, Mitsumoto H, Chung W, Croft GF, et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science. 2008;321(5893):1218-1221.
doi pubmed
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Neurology Research is published by Elmer Press Inc.