







| Journal of Neurology Research, ISSN 1923-2845 print, 1923-2853 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, J Neurol Res and Elmer Press Inc |
| Journal website http://www.neurores.org |
Original Article
Volume 2, Number 2, April 2012, pages 39-43
Levetiracetam Protects Spinal Motor Neurons Against Glutamate-Induced Neurotoxicity in Culture
Yasuhiro Yoshiia, Ken Ikedaa, b, Yasuo Iwasakia
aDepartment of Neurology, Toho University Omori Medical Center, 6-11-1 Omorinishi, Otaku, Tokyo, 143-8541, Japan
bCorresponding author: Ken Ikeda
Manuscript accepted for publication April 18, 2012
Short title: Levetiracetam Protects Spinal Motor Neurons
doi: https://doi.org/10.4021/jnr89w
| Abstract | ▴Top |
Background: Levetiracetam is widely used in numerous patients with epileptic seizures. In an animal model of cerebral ischemia after the occlusion of the internal carotid artery, pre-treatment with levetiracetam could reduce the infarct size. However, little is known how this antiepileptic drug can act on motor neurons. The purpose of this study is to evaluate whether levetiracetam has neuroprotective effects on spinal motor neurons in glutamate-treated culture of neonatal rats.
Methods: Postnatal organotypic spinal cord cultures were exposed to glutamate (10-5 M) alone or glutamate (10-5 M) plus levetiracetam (10-5, 10-6 and 10-7 M). Cultures were treated for two weeks and morphological changes were examined. The number of surviving spinal motor neurons and the activities of choline acetyltransferase (ChAT) were measured.
Results: Cultures treated with glutamate had significant reductions of the surviving motor neurons and the ChAT activities compared to non-glutamate-treated control culture. Cultures added both glutamate and levetiracetam showed significantly suppression of the neuronal loss and potentiation of the ChAT activities.
Conclusions: The present study indicated that levetiracetam prolonged the survival and the function of spinal motor neurons against glutamate-induced neurotoxicity in culture. This drug may have a therapeutic potential for several diseases that kill or degenerate the spinal motor neurons, including spinal cord injury and amyotrophic lateral sclerosis.
Keywords: Glutamate-induced neurotoxicity; Neuroprotection; Levetiracetam; Spinal motor neuron; Choline acetyltransferase; Amyotrophic lateral sclerosis
| Introduction | ▴Top |
A previous study of post-traumatic brain damage has shown a potent action of N-methyl-D-aspartate (NMDA) antagonists in the neuronal death associated with cerebral ischemia [1]. Neurological dysfunction or neuropathological changes involved in glutamate release can contribute to numerous pathophysiological conditions mediated by calcium overload in both neurons and glial cells [2]. Glutamate-induced neurotoxicity might reveal neuronal damage in ischemic stroke and neurodegenerative disorders, such as Alzheimer disease, Parkinson’s disease and amyotrophic lateral sclerosis (ALS) [3]. Kainic acid is known as an analogue of glutamate. This substance produces convulsions by the activation of NMDA receptors that enters calcium into cells, leading to cell death process [4]. Several antioxidant agents can prevent the excitotoxicity induced by glutamate or kainic acid [5]. Otherwise, ALS is a fatal disease with muscular weakness and respiratory failure caused by selective degeneration of upper and lower motor neurons. Neuronal glutamate excitotoxicity may be hypothesized as one of possible mechanisms in this neurodegenerative disease [6]. Riluzole, an inhibition of glutamate release, is the only drug that has been approved for the treatment of ALS [7]. Levetiracetam (LEV), (S)-alpha-ethyl-2-oxo-pyrrolidine acetamide, is widely used as an antiepileptic drug. LEV has antiepileptic effects in rodent models of epilepsy and seizures [8, 9]. This drug also has neuroprotective effects in cerebrovascular ischemia models [10]. However, little is known about effects of LEV on motor neurons. Herein we aimed to study whether this drug can save spinal motor neurons against glutamate-induced neurotoxicity in culture.
| Material and Methods | ▴Top |
Organotypic culture of spinal cord
Organotypic spinal cord was performed from the lumbar spinal cord of 10-day-old Sprague-Dawley rats (Sanhkyo Laboratories, Tokyo, Japan). The lumber spinal cords were removed and spliced into 5 mm thickness of transverse section. Each one slice was placed on Falcon dish (30 mm in diameter) with a milipore CM semipermeable membrane insert in Dulbecco’s modified Eagle’s medium of 3 mL. Antibiotics and antifungal agents were not used. Cultures were incubated at 37 °C in 5% CO2 and 95% air. Purified LEV was supplied from Otsuka Pharmaceutical Co. Ltd., Tokyo, Japan. To assess neuroprotective effects of LEV against glutamate-induced neurotoxicity, cultures were incubated for two weeks with glutamate (10-5 M) or glutamate (10-5 M) plus three doses of LEV (10-5, 10-6 and 10-7 M). Culture medium alone and administration of glutamate or LEV were exchanged twice weekly. Finally, five experimental groups consisted of the untreated control culture, the glutamate-treated culture, and the glutamate + three doses of LEV-treated culture.
Morphological assessment
The gross morphology of the culture was monitored daily by inverted phase-contrast microscope (Nikon Co., TMD-1S, Tokyo, Japan). After 2 weeks incubation, specimen of cultured spinal cord were separated gently and immersed in 0.1 M phosphate buffer and 4% paraformaldehyde, pH 7.4 for 2 hours. The tissues were dehydrated and embedded in paraffin, and serial transverse sections (6 µm thickness) were made. Every fifth section (6 µm thickness; 24 µm interval) was collected, deparaffinized and stained with cresyl violet (Nissl staining). The number of motor neurons larger than 25 µm in a diameter and with at least one thick process as defined by the criteria of Bilak et al [11] in the ventral half of the gray matter was counted under light microscope. In the control cultures (n = 10) and each experimental culture (n = 10), Nissl staining was performed on 30 slices per group. Two investigators who were unaware of treatment status determined the number of motor neurons. The number of motor neurons per slice was then calculated.
ChAT activities of cultured spinal cord
Choline acetyltransferase (ChAT) activity was a reliable surrogate biochemical marker of the motor neuron. According to a previously described method [12], ChAT activity was measured at 2 weeks after initial planting of the organotypic lumbar cord culture. For the ChAT assay, cultures were treated with a mixture containing 0.2 mM (14C) acetyl CoA (Amersham, Tokyo, Japan), 50 mM sodium phosphate buffer (pH 7.4), 300 mM NaCl, 8 mM choline chloride, 20 mM EDTA, and 0.1 mM physostigmine. The homogenates (2 µL) were put in microtubes, mixed, and incubated for 20 minutes at 37 °C. The reaction was stopped by adding 5 mL of 10 mM sodium phosphate buffer (pH 7.4); the contents were transferred to scintillation vials. Then, 2 mL acetonitrile containing 10 mg of sodium tetraphenylboron, and 10 mL of toluene scintillation mixture (0.4% DPO, 0.01% POPOP) were added to the vials. The ChAT of cultures and (14C) acetyl CoA were reacted, leading to production of (14C) acetylcholine. The (14C) acetylcholine was fluorescent in the toluene scintillation mixture; level of fluorescence was calculated by using a liquid scintillation counter (Aloka LC-3500, Tokyo, Japan). The protein concentration was determined by using a Bio-Rad protein assay kit (Bio-Rad Laboratories, Tokyo, Japan) with bovine serum albumin as standard. The enzyme activity was expressed as acetylcholineproduction per mg protein per minute. Ten cultures from each experimental group (n =10/group) were analyzed.
Statistical analysis
Data were expressed as the mean (SD) in all groups. Statistical analysis was done using one-way analysis of variance followed by Scheffe’s test. The significance level was set at 0.05 in both tests.
| Results | ▴Top |
Glutamate-treated cultures decreased the number of motor neurons at 80% compared to untreated control culture. LEV-treated cultures significantly inhibited the loss of motor neurons (Fig. 1). LEV (10-6 and 10-5 M) treatment attenuated the loss of motor neurons compared to glutamate-treated cultures (Fig. 2). There was no significant difference for the surviving motor neurons between the glutamate group and the glutamate + LEV (10-7 M) group. The ChAT activity in glutamate-treated cultures was decreased approximately 50% in compared to untreated control cultures. In cultures treated with LEV of 10-6 and 10-5 M, ChAT activity was enhanced (Fig. 3).
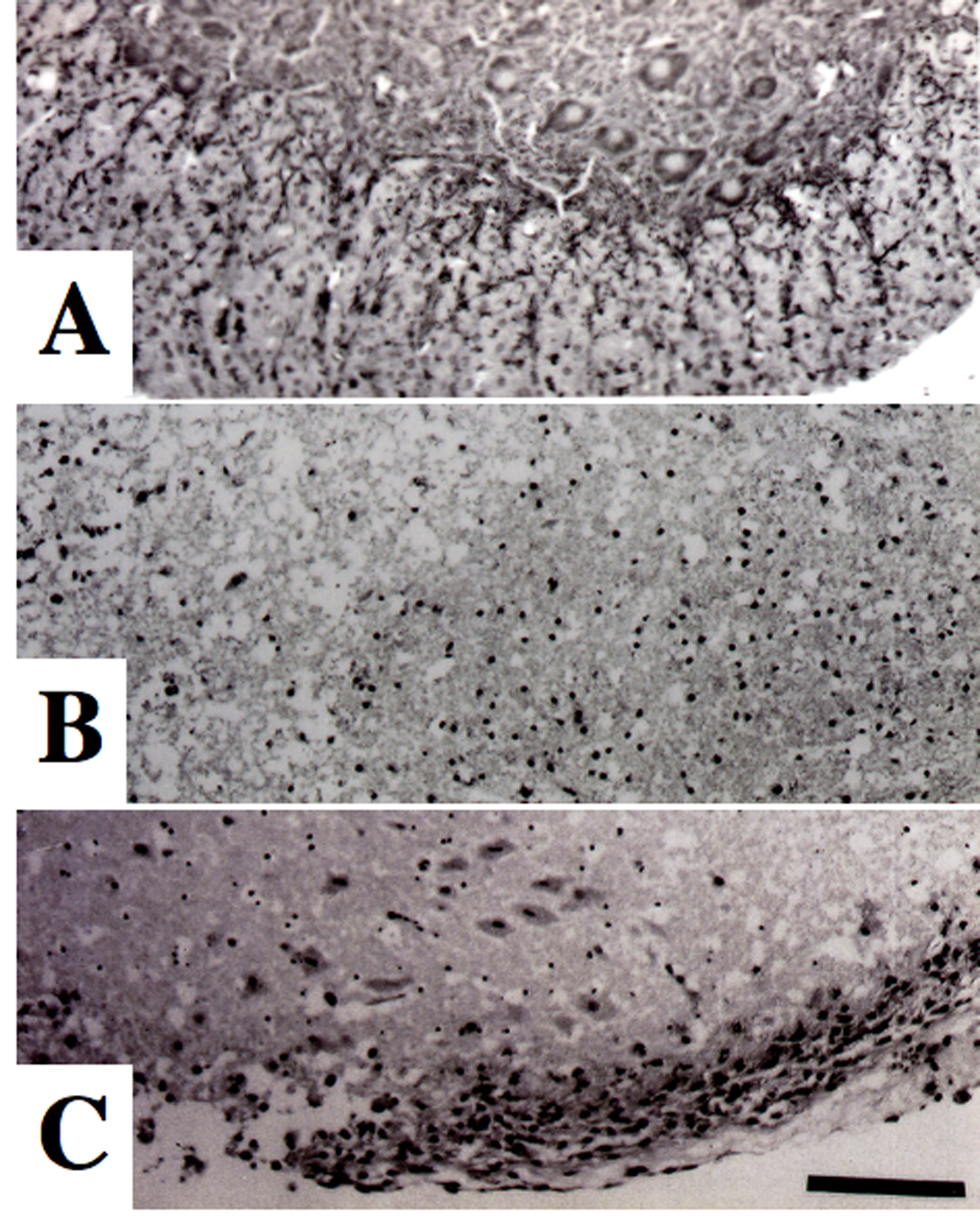 Click for large image | Figure 1. Morphological changes of spinal motor neurons on Nissl staining. (A) Control culture. (B) Glutamate-treated culture. (C) Culture treated with glutamate and levetiracetam (10-5 M). Glutamate administration produced a marked loss of motor neurons compared to control culture. Co-administration of levetiracetam attenuated the loss of motor neurons in glutamate-treated culture. Bar = 100 µm. |
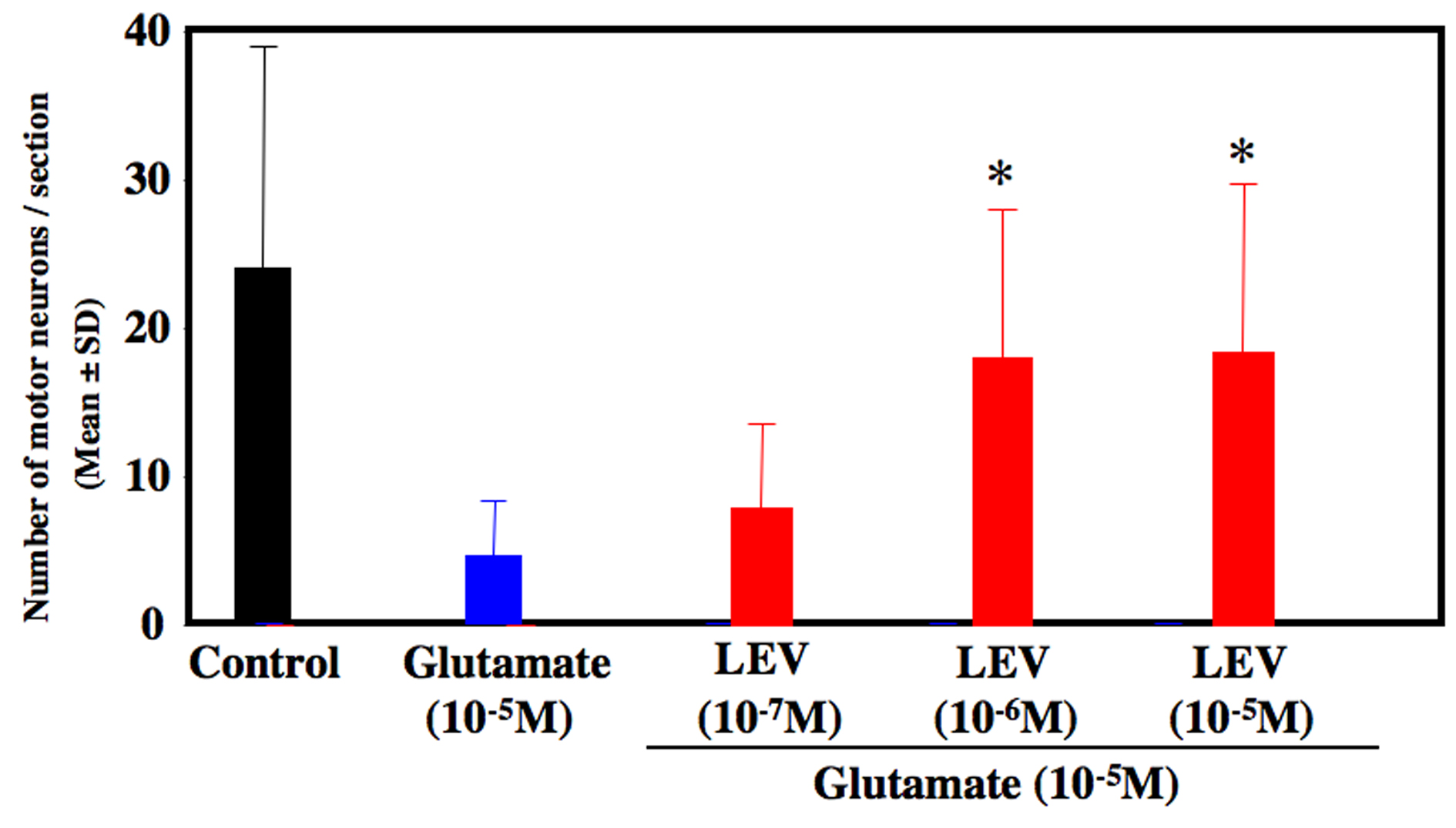 Click for large image | Figure 2. Number of surviving motor neurons. After 2 weeks of cultures, the number of motor neurons was decreased approximately 80 % in glutamate-treated culture compared to control culture. Levetiracetam treatment (10-6 and 10-5 M) inhibited the loss of motor neurons. *P < 0.05 between the glutamate group and the glutamate + levetiracetam group (10-6 and 10-5 M) by Scheffe’s test. |
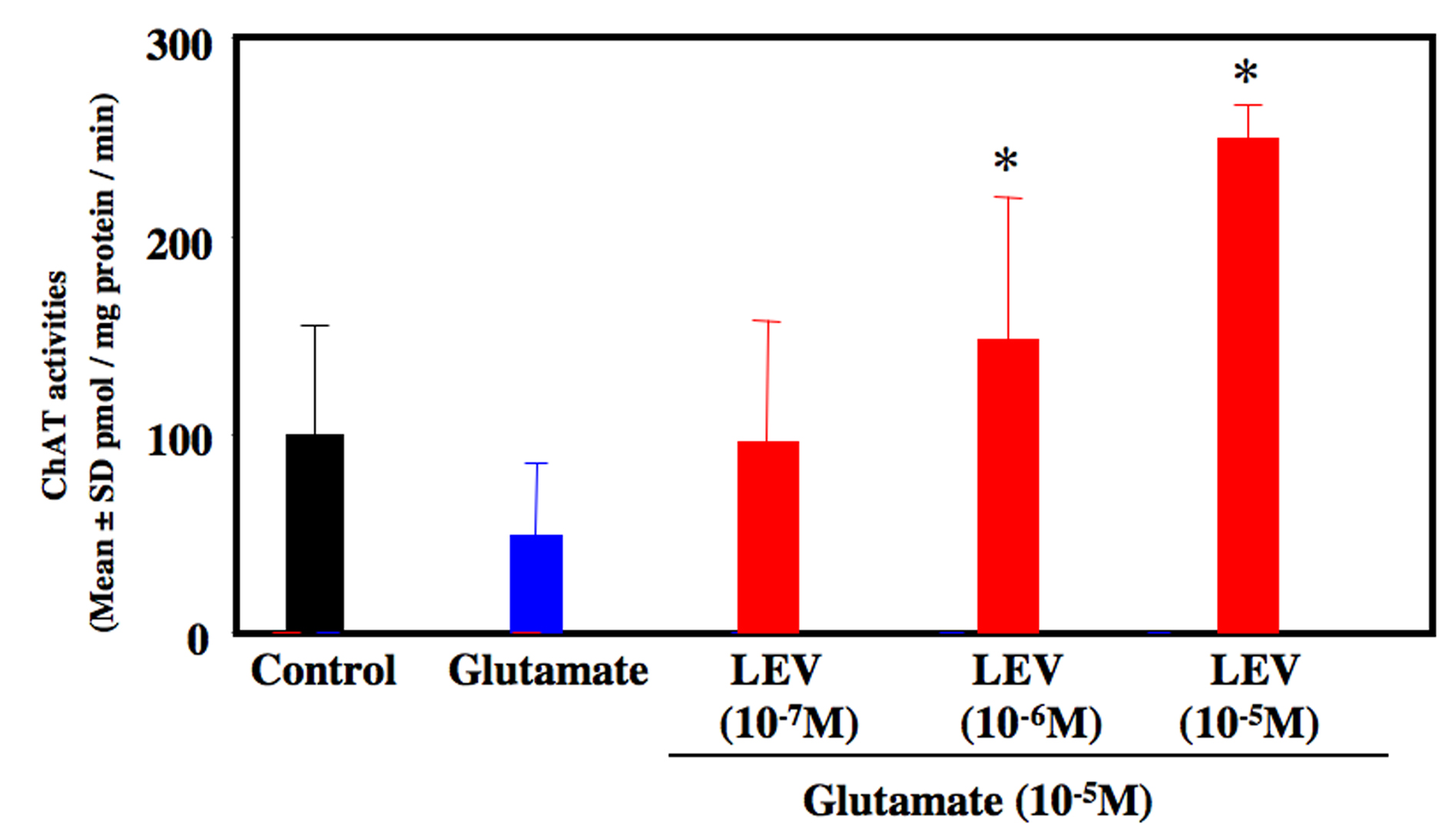 Click for large image | Figure 3. ChAT activity of organotypic spinal cord culture. Administration of glutamate reduced ChAT activities approximately 50 % compared to control culture. Co-administration of levetiracetam (10-6 and 10-5 M) increased ChAT activity compared to glutamate-treated culture. *P < 0.05 between the glutamate group and the glutamate + levetiracetam (10-6 and 10-5 M) group by Scheffe’s test. |
| Discussion | ▴Top |
In this model, we used two independent methods to show neuroprotective effects of LEV on cultured motor neurons. At first, we evaluated the number of surviving motor neurons in organotypic cultures. Second we measured the ChAT activity, because ChAT is a reliable marker mainly restricted to motor neurons in the spinal cord. The present study suggested neuroprotective effects of LEV on spinal motor neurons by glutamate-induced neurotoxicity. The morphological and biochemical results showed dissimilar degree of neuroprotective effects. ChAT activity was potentated in cultures treated with glutamate plus LEV (10-6 and 10-5 M) compared to control cultures. Therefore, LEV-treated cultures might enhance biochemical compartments more than morphological counterparts. Glutamate neurotoxicity can be mediated by both NMDA and non-NMDA glutamate receptors [13]. Glutamate may induce various neurotoxic cellular processes, such as formation of oxygen free radicals and calcium release. An increased production of glutamate represents a key established pathogenetic mechanism leading to neuronal death [3]. As consequence, the main therapeutic approaches include agents able to inhibit the glutamate pathology [14]. The mechanisms underlying the neuroprotective effects of LEV are not fully understood. LEV has the ability to block Ca2+ influx into the neuronal cells. This effect is reasonable to consider possible activity to oxidative stress and the consequently inflammatory response [15]. Another mechanism for the suppression of glutamate overflow is the inhibitory modulation of synaptic vesicle protein 2A (SV2A) receptor. SV2A-knockout mice show seizure. Without SV2A, presynaptic calcium accumulation during repetitive stimulation causes abnormal increases in the neurotransmitter over flow. Therefore, the binding of the SV2A receptor with LEV might restore the ability of neurons to reduce the excessive glutamate overflow [16, 17]. It has been demonstrated that deleterious consequence of oxidative damage is the triggering of inflammatory cytokines able to amplify the neuronal damage. Tumor necrosis factor alpha, interleukin-6, and interleukin-1b (IL-1b) have been shown to cause neuronal damage and are also involved in the pathogenetic mechanisms of neurodegenerative diseases [18]. IL-1b is markedly increased during cerebral ischemia and other models of neurotoxicity [19, 20]. LEV has been reported to reduce the IL-1b expression markedly [21]. Among the inflammatory cytokines in the brain injury, IL-1b seems to be an important factor. Moreover, LEV stimulates the expression of brain-derived neurotrophic factor (BDNF) on rat cortical astrocyte cultures [22]. BDNF is well known to enhance neuronal growth and survival in spinal motor neurons [23, 24]. Riluzole is a drug that inhibits glutamate release and increase survival of both ALS patients [9] and SOD1 transgenic mice [25]. Spinal neurons possess noradrenalin and other transmitter system, in addition to the cholinergic system. Whether glutamate-induced toxicity is specific for the cholinergic system remains unclear. Further studies are needed to elucidate the mechanism underlying the neuroprotection of LEV against glutamate toxicity-triggered death of spinal motor neurons. Finally, LEV may have therapeutic potential in patients with motor neuron damage, such as ALS and traumatic spinal cord.
| References | ▴Top |
- Bullock R, Kuroda Y, Teasdale GM, McCulloch J. Prevention of post-traumatic excitotoxic brain damage with NMDA antagonist drugs: a new strategy for the nineties. Acta Neurochir Suppl (Wien). 1992;55:49-55.
pubmed - Meldrum BS. Glutamate as a neurotransmitter in the brain: review of physiology and pathology. J Nutr. 2000;130(4S Suppl):1007S-1015S.
pubmed - Doble A. The role of excitotoxicity in neurodegenerative disease: implications for therapy. Pharmacol Ther. 1999;81(3):163-221.
pubmed doi - Turski L. [The N-methyl-D-aspartate receptor complex. Various sites of regulation and clinical consequences]. Arzneimittelforschung. 1990;40(5):511-514.
pubmed - Kim HC, Jhoo WK, Bing G, Shin EJ, Wie MB, Kim WK, Ko KH. Phenidone prevents kainate-induced neurotoxicity via antioxidant mechanisms. Brain Res. 2000;874(1):15-23.
pubmed doi - Lipton SA, Rosenberg PA. Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med. 1994;330(9):613-622.
pubmed doi - Lacomblez L, Bensimon G, Leigh PN, Guillet P, Meininger V. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study Group II. Lancet. 1996;347(9013):1425-1431.
pubmed - Klitgaard H, Matagne A, Gobert J, Wulfert E. Evidence for a unique profile of levetiracetam in rodent models of seizures and epilepsy. Eur J Pharmacol. 1998;353(2-3):191-206.
pubmed doi - Loscher W, Honack D, Rundfeldt C. Antiepileptogenic effects of the novel anticonvulsant levetiracetam (ucb L059) in the kindling model of temporal lobe epilepsy. J Pharmacol Exp Ther. 1998;284(2):474-479.
pubmed - Hanon E, Klitgaard H. Neuroprotective properties of the novel antiepileptic drug levetiracetam in the rat middle cerebral artery occlusion model of focal cerebral ischemia. Seizure. 2001;10(4):287-293.
pubmed doi - Bilak MM, Corse AM, Bilak SR, Lehar M, Tombran-Tink J, Kuncl RW. Pigment epithelium-derived factor (PEDF) protects motor neurons from chronic glutamate-mediated neurodegeneration. J Neuropathol Exp Neurol. 1999;58(7):719-728.
pubmed doi - Iwasaki Y, Ichikawa Y, Igarashi O, Ikeda K, Konno S, Aoyagi J, Kinoshita M. Temocapril prevents motor neuron damage and upregulation of cyclooxygenase-II in glutamate-induced neurotoxicity. Neurol Res. 2003;25(3):301-304.
pubmed doi - Shaw PJ. Excitotoxicity and motor neurone disease: a review of the evidence. J Neurol Sci. 1994;124 Suppl:6-13.
pubmed - McCulloch J. Excitatory amino acid antagonists and their potential for the treatment of ischaemic brain damage in man. Br J Clin Pharmacol. 1992;34(2):106-114.
pubmed - Lukyanetz EA, Shkryl VM, Kostyuk PG. Selective blockade of N-type calcium channels by levetiracetam. Epilepsia. 2002;43(1):9-18.
pubmed doi - Stahl SM. Psychopharmacology of anticonvulsants: levetiracetam as a synaptic vesicle protein modulator. J Clin Psychiatry. 2004;65(9):1162-1163.
pubmed doi - Lynch BA, Lambeng N, Nocka K, Kensel-Hammes P, Bajjalieh SM, Matagne A, Fuks B. The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc Natl Acad Sci U S A. 2004;101(26):9861-9866.
pubmed doi - Eriksson C, Van Dam AM, Lucassen PJ, Bol JG, Winblad B, Schultzberg M. Immunohistochemical localization of interleukin-1beta, interleukin-1 receptor antagonist and interleukin-1beta converting enzyme/caspase-1 in the rat brain after peripheral administration of kainic acid. Neuroscience. 1999;93(3):915-930.
pubmed doi - Wang CX, Shuaib A. Involvement of inflammatory cytokines in central nervous system injury. Prog Neurobiol. 2002;67(2):161-172.
pubmed doi - Boutin H, LeFeuvre RA, Horai R, Asano M, Iwakura Y, Rothwell NJ. Role of IL-1alpha and IL-1beta in ischemic brain damage. J Neurosci. 2001;21(15):5528-5534.
pubmed - Marini H, Costa C, Passaniti M, Esposito M, Campo GM, Ientile R, Adamo EB, et al. Levetiracetam protects against kainic acid-induced toxicity. Life Sci. 2004;74(10):1253-1264.
pubmed doi - Cardile V, Pavone A, Gulino R, Renis M, Scifo C, Perciavalle V. Expression of brain-derived neurotrophic factor (BDNF) and inducible nitric oxide synthase (iNOS) in rat astrocyte cultures treated with Levetiracetam. Brain Res. 2003;976(2):227-233.
pubmed doi - Iwasaki Y, Ikeda K, Kinoshita M. Molecular and cellular mechanism of glutamate receptors in relation to amyotrophic lateral sclerosis. Curr Drug Targets CNS Neurol Disord. 2002;1(5):511-518.
pubmed doi - Mitsumoto H, Ikeda K, Klinkosz B, Cedarbaum JM, Wong V, Lindsay RM. Arrest of motor neuron disease in wobbler mice cotreated with CNTF and BDNF. Science. 1994;265(5175):1107-1110.
pubmed doi - Gurney ME, Cutting FB, Zhai P, Doble A, Taylor CP, Andrus PK, Hall ED. Benefit of vitamin E, riluzole, and gabapentin in a transgenic model of familial amyotrophic lateral sclerosis. Ann Neurol. 1996;39(2):147-157.
pubmed doi
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Journal of Neurology Research is published by Elmer Press Inc.